A Case Series and Review of CDKL5 Deficiency Disorder: More than Rett
A Case Series and Review of CDKL5 Deficiency Disorder: More than Rett
A B S T R A C T
CDKL5 Deficiency Disorder (CDD) or CDKL5-associated epileptic encephalopathy (CAEE) is a distinct X-linked dominant epileptic encephalopathy that shares many features with Rett syndrome. In the past decade, mutations in CDKL5 gene were identified as part of the molecular heterogeneity of MECP2-negative Rett syndrome. CDD has increasingly gained recognition as a distinct molecular and clinical phenotype. Here we present four new patients with CDD: one with a clinical presentation of reflex seizures previously reported as a case report by the same group, and three cases with novel disease-causing mutations. The emerging distinct phenotype of CDD should allow the clinician to suspect the diagnosis early and avoid a lengthy diagnostic odyssey.
Keywords
CDKL5, Rett, epileptic encephalopathy, infantile seizures, microcephaly, spasms, CDD
Introduction
CDKL5-associated epileptic encephalopathy (CAEE) or CDKL5 Deficiency Disorder (CDD) has been increasingly recognized in the last decade as a distinct clinical entity [1, 2]. CDD or Early Infantile Epileptic Encephalopathy, Type 2 (OMIM # 300672) usually presents in early infancy with seizures, severe development delay, loss of milestones and postnatal microcephaly [3]. CAEE or CDD was originally described by several groups in 2004-2005 [4-7]. This was motivated in large part by the heterogeneity of the loose grouping of diseases referred to alternately as Rett syndrome (RTT), atypical Rett syndrome, variant Rett syndrome or Rett-like syndromes. The Rett name has come to refer to three essential characteristics: postnatal deceleration of head growth, loss of acquired spoken language or purposeful hand skills and gait abnormalities [8].
In patients with classical RTT, about 20% are found to be MECP2 mutation-negative, and even fewer MECP2 mutations are found in patients with atypical and variant RTT [9]. Mutations in CDKL5, FOXG1, ANKRD31, STXBP1 and CHRNA5 have been identified in some MECP2-negative patients [8, 10, 11].
The CDKL5 gene is located on Xp22.13. It is approximately 240 kb long and contains 24 exons, with only exons 2-21 translated. It encodes a 118 kD protein. There are at least three distinct human isoforms with different C-terminus regions that are widespread in tissues including the brain, but it is unknown whether they have distinct functions [12]. CDKL5 is expressed throughout the body, especially in the brain, testes and thymus. In the brain, it first appears in neurons that have migrated to their final position in the cerebral cortex between 12-20 weeks of gestation [13, 14]. CDKL5 was formerly known as STK9, or serine-threonine kinase 9, due to its highly conserved serine-threonine kinase domain near the N-terminus [15].
As more knowledge is gained about CDKL5 and its role in neurodevelopment, there has been a movement to recognize CDD as a distinct clinical entity instead of just part of the genetic heterogeneity of Rett syndrome. In recent publications and guidelines, there is some evidence for CDKL5 primary candidate gene testing and it is recommended in certain contexts [4, 16-21]. Recently, with the move towards diagnostic testing through gene panels and clinical exome sequencing, it is now possible to screen for mutations in many genes at the same time. Nonetheless, CDD is an important diagnosis to suspect early on in order to expedite molecular confirmation and avoid a lengthy diagnostic odyssey. In this paper, we present a case-series of four patients including a description of one with an unusual seizure type and three others with novel mutations to illustrate our local institutional experience with CDD. We emphasize the unique features of the disease that should lead the clinician to suspect CDD first and foremost.
Table 1: Patients’ clinical features.
|
|
Patient 1 - “Reflex seizures” |
Patient 2 – “Novel frameshift” |
Patient 3 – “Novel splice site” |
Patient 4 – “Novel nonsense” |
|
Seizure onset |
6 months |
2 months |
2 months |
3 months |
|
Sex |
Female |
Female |
Female |
Female |
|
Family history |
Unremarkable |
Unremarkable |
Unremarkable |
Unremarkable |
|
Seizure semiology |
6 months-18 months: startle spasms sometimes progressing to GTC only with bathing 18 months: tonic-spasms sequence sometimes progressing to GTC spontaneous or startle to submersion in water and loud noise. No hypermotor component. |
2 months-2.5 years: Tonic, focal, autonomic symptoms 2.5 years: seizures in clusters |
2 months-10 months: focal-tonic 10 months: hypermotor-tonic-spasm sequence |
3 months-12 months: GTC 12 months-1.5 years: hypermotor-tonic-spasm sequence 1.5 years-2 years: tonic-spasm 2 years: GTC, focal |
|
EEG findings |
8 months: Excess high amplitude background slowing, somewhat poorly developed sleep features and parietal region hemispheric asymmetry 2 years: Multiple independent spike foci (MISF) and slow background activity for age |
Delta slowing of the background followed by generalized attenuation during seizures and multifocal interictal epileptiform abnormalities |
Bilateral independent frontal interictal discharges during sleep. During ictal event, there was high amplitude 2 Hz delta (hypermotor), followed by diffused attenuation (tonic) and spasms complex afterward. (refer to Figure 1.). |
Bilateral independent frontal interictal discharges during sleep. No EEG of ictal events was ever captured. |
|
Development |
Severe delayed gross motor, fine motor, language and social |
Severe delayed gross motor, fine motor, language and social |
Severe delayed gross motor, fine motor and language Mild delay social |
Severe delayed gross motor, fine motor, language and social |
|
Stereotypies |
Abnormal hand movements to midline |
Hyperoral Hand flapping |
Abnormal hand movements to midline |
Abnormal hand movements to midline Obsession with hands |
|
Systemic disease |
GERD |
None |
None |
None
|
|
Physical examination |
Normal growth parameters Axial hypotonia, hyperreflexia Poor tracking/eye contact |
Microcephaly (<2 SD) Axial hypotonia |
Microcephaly (<2 SD) Axial hypotonia Intermittent dysconjugate strabismus |
HC decreased growth velocity Axial hypotonia |
|
Investigations |
NBS, lactate, uric acid, ammonia, plasma and urine amino acids, urine organic acids, succinylpurines, Batten disease screen of CLN1, CLN2 and ultrastructural examination for membrane bound inclusions, acylcarnitine profile, alpha galactosidase, chitotriosidase, galactocerebrosidase, urine and plasma creatine and guanidinoacetate, TIEF, lysosomal enzyme activities, palmitate oxidation and ATP synthesis, CSF analysis (neurotransmitters, lactate, glucose, protein, amino acids), MRI-brain with spectroscopy, array-CGH, MECP2 sequencing, CDKL5 MLPA and array-CGH |
NBS, lactate, lipoprotein profile, plasma and urine amino acids, urine organic acids, acylcarnitine profile, total and free serum carnitine levels, plasma ammonia, total plasma homocysteine, serum CK, liver enzymes, urine alpha-AASA, creatine, biotinidase, VLCFA, Batten disease screen, CSF analysis (amino acids, lactate, glucose, protein, cell count, neurotransmitters), MRI-brain with spectroscopy, karyotype, gene sequencing (SCN1A, MECP2, CDKL5) |
NBS, plasma and urine amino acids, total and free carnitine, acylcarnitine, urine organic acids, CBC, electrolytes, Ca, Mg, phosphate, lipase, liver enzymes, albumin, lactate, , uric acid, ammonium, VLCFA, homocysteine, CK, lysosomal enzyme activity, Krabbe, CLN1, CLN2, plasma pipecolic acid, CSF analysis (protein, glucose, lactate, neurotransmitters, pyridoxal 5’ phosphate, 5-HIAA, HVA), Vit E, Vit B12, Vit B6, urine methylmalonic acid, cholesterol, MRI-brain with spectroscopy, array-CGH, gene sequencing (PNPO, CDKL5) |
NBS, plasma and urine amino acids, urine organic acids, lactate, total and free carnitine, TIEF, creatine, GAA, sialic acid, lipase, CBC, electrolytes, Ca, Mg, phosphate, lipoproteins, liver enzymes, albumin, lactate, uric acid, ammonia, LCFA, VLCFA, homocysteine, CK, MRI-brain with spectroscopy, array-CGH, gene sequencing (PNPO, MECP2, CDKL5) |
|
Molecular diagnosis |
c.162_99del261 |
c.2480_2486dupCAGATCT |
c.99+1G>A |
c.1238C>A |
|
Age at diagnosis |
2 years |
2 years |
<1 year |
2 years |
|
Antiepileptics |
Phenobarbital, Topirimate, Clobazam, Valproate |
Phenobarbital, Topirimate, Clobazam, Valproate, Keppra, Vitamin B6, Phenytoin, Lamotrigine, Nitrazepam, Oxcarbazepine, Mirtazipine, KCI, Levetiracetam, ketogenic diet |
Phenobarbital, Topirimate, Clobazam, Levetiracetam, Lamotrigine, Vit B6, Vit B12, folate, ketogenic diet |
Phenobarbital, Topirimate, Clobazam, Valproate, Levetiracetam, Oxcarbazepine, Gabapentin |
|
Other medications |
Ranitidine |
Carnitine |
None |
None |
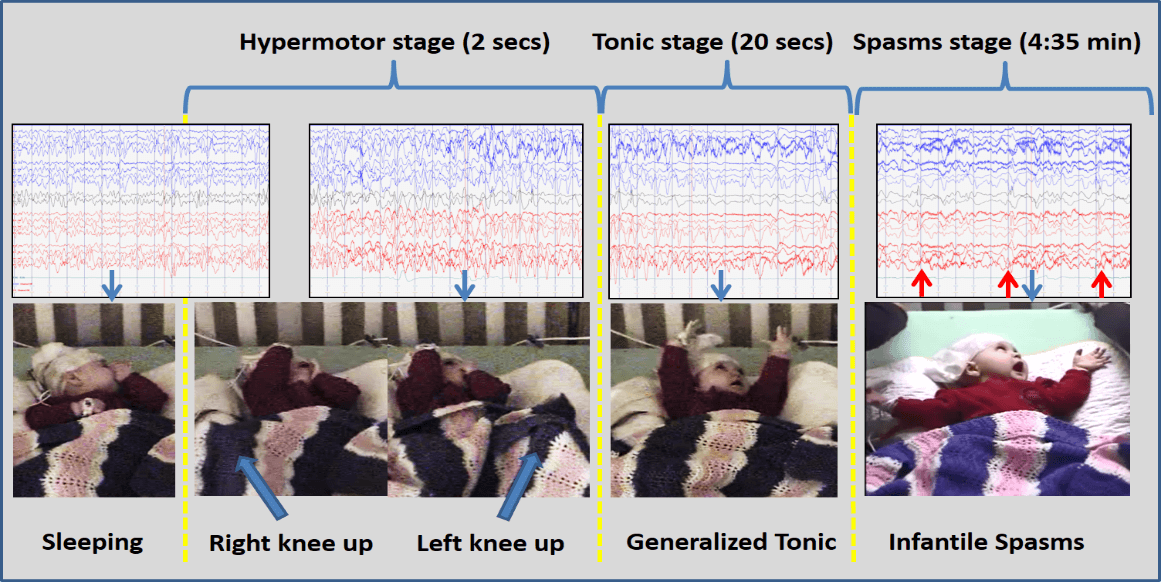
Figure 1: Patient 3 with pathognomonic hypermotor-tonic-spasms sequence of CDD. EEG settings: APB montage, LFF 1Hz, HHF 70Hz, Notch 60Hz, Sensitivity 7uV/mm (but spasms sample 15 uV/mm to better appreciate trace), paper-speed 30 mm/sec. Time in brackets represent length of each seizure type. Blue arrows on the EEG trace indicated the actual moment of the below shown picture and the red arrows indicated the spasms complex on the EEG.
Illustrative Case Summaries
I Clinical Features
The four patients reported in this paper all presented with severe neurodevelopmental disorders and seizures with their features are summarized in (Table 1). An in-depth clinical description of patient 1 reflects her unusual presentation with reflex seizures, which to our knowledge has only been reported once with MECP2-related disease and never with CAEE and twice with CDD including patient 1 [22-24].
i Patient 1
The patient was born at term weighing 3.44 kg by emergency Cesarean section due to failure to progress and fetal distress. She had normal growth in early infancy but there were concerns about developmental delay as by 6 months old she was not sitting and did not have purposeful hand use. She also developed reflex seizures upon bathing and developed short startle spasms and spontaneous spasm-like seizures in later infancy. The first event was witnessed two days after receiving her 6 month immunizations: she was being bathed by her mother when she turned her head to the right with a fixed stare then her whole body went stiff followed by generalized tonic-clonic movements. The entire episode lasted about 30 seconds and the patient returned to normal afterwards. Another event occurred later that night when bathing with the same pattern and duration. Subsequently, similar episodes occurred lasting 30-60 seconds, and only during bathing. The frequency decreased as the parents were advised to limit full bathing. At 1.5 years old, startle episodes were witnessed with loud noises and submersion in water, involving bilateral upper limb hyperextension raised above the head with both eyes wide open, lasting 1-3 seconds. At 20 months old, spontaneous generalized brief tonic seizures followed by hyperextensive-spasm-like seizures sometimes progressing to generalized tonic-clonic seizures were observed, at a frequency of about 2-3 per week. Each seizure lasted around 30-45 seconds.
At 12 months of age, rhythmic repetitive midline hand wringing movements started. By 20 months of age she was significantly developmentally delayed with language at a 3-4 month level. Family history was significant for a maternal uncle with febrile seizures and a maternal cousin with muscular dystrophy. There was no reported consanguinity. On clinical examination, the patient was non-dysmorphic, alert but irritable. She was axially hypotonic and visual interest and tracking were poor. Sensation, peripheral tone and strength were normal. Deep tendon reflexes were normal. There were no cutaneous abnormalities. Cardiorespiratory and abdominal examinations were normal.
On subsequent serial evaluations, there was no deceleration of head growth: her weight, height and head circumference all followed between the 25-50th %ile throughout the evolution of her disease. At 14 months of age, visual tracking improved. At 20 months of age, axial hypotonia had improved, however she had become hyperreflexic. Electroencephalogram (EEG) performed at 8 months of age showed excess high amplitude background slowing, somewhat poorly developed sleep features and parietal region hemispheric asymmetry, overall indicating diffuse brain dysfunction. Repeat EEG at 22 months old showed multiple independent spike foci (MISF) and slow background activity for age, indicating epileptic encephalopathy. Brain-MRI showed normal myelination for age with normal MR spectroscopy.
Metabolic investigations and CSF analysis were negative. Oligonucleotide array-CGH analysis was normal. Based on a suspicion of Rett Syndrome or CAEE, further tests were performed. Sequence analysis of MECP2 was normal. Targeted array-CGH with exon-level resolution of CDKL5 was carried out (GeneDx, Gaithersburg, MD) which detected a deletion encompassing exons 2-3, confirmed by sequence analysis. She was started on phenobarbital but experienced breakthrough seizures. Various combinations of topiramate, valproic acid and clobazam were added with a seizure frequency of approximately of 2-3 per week. Ketogenic diet was offered but declined. Her development has remained poor.
ii Patients 2, 3 & 4
The clinical histories and investigations performed in these patients are summarized in (Table 1). These three patients all showed abnormal development from birth with complex seizure disorders beginning in infancy and stereotypic hand movements.
II Molecular Findings
i Patient 1
CDKL5 targeted array-CGH with exon-level resolution confirmed by sequence analysis detected a deletion encompassing exons 2-3, c.162_99del261 (GeneDx, Gaithersburg, MD). This has been previously reported and results in a premature stop codon, a known pathogenic mutation [25].
ii Patient 2
CDKL5 gene sequencing detected a de novo duplication in exon 17, c.2480_2486dupCAGATCT, resulting in a frameshift (Boston University School of Medicine, Center for Human Genetics, Boston, MA). This is a novel change that has not been reported before in ExAC [26]. Only pathogenic point mutations in exon 17 have previously been reported [20]. In a patient with a previously reported frameshift mutation in exon 18, a truncated CDKL5 transcript was detected [6]. A truncated protein would lack the C-terminus and would not localize correctly in the cell, as demonstrated in vitro [27]. Accordingly, any reading frame altering mutations proximal to exon 18 are null-variants. Therefore, this mutation is classified as pathogenic according to ACMG criteria [28].
iii Patient 3
CDKL5 gene sequencing detected a de novo donor splice site mutation in intron 3, c.99+1G>A (Boston University School of Medicine, Center for Human Genetics, Boston, MA). The mutation in our patient is a novel change that has not been reported before in ExAC [26]. A similar mutation was previously reported at the same location (c.99+1G>T) with in vitro evidence that it alters splicing and results in a frameshift and premature stop codon, producing a transcript lacking exon 3 [20]. Since the novel change disrupts an established canonical splice site, it is classified as pathogenic according to ACMG criteria [28].
iv Patient 4
CDKL5 gene sequencing detected a de novo point mutation in exon 12, c.1238C>A (Boston University School of Medicine, Center for Human Genetics, Boston, MA). The mutation in our patient is a novel change that has not been reported before in ExAC [26]. A nonsense mutation at the same location (c.1238C>G) was previously reported [29]. Similar to other truncating mutations that abolish the C-terminus, this product would not localize correctly in the cell, as demonstrated in vitro [27]. Therefore, it is classified as pathogenic according to ACMG criteria [28].
Discussion
I Clinical Features
CDD has been historically characterized by seizures and Rett-like features that emerge in the first year of life and may even satisfy criteria for atypical RTT [8]. Since the first cohorts of patients tested were derived from MECP2-negative RTT populations, subsequent broader offering of testing and identification of affected family members have revealed much clinical heterogeneity. In one cohort of patients with early seizures and encephalopathy, the yield of CDKL5 mutations found in patients who satisfied criteria for atypical RTT was almost the same as those who did not satisfy criteria (about 13% and 10% respectively) [20].
CDD can have wide phenotypic variability, from early-onset severe epileptic encephalopathy with no period of normal development, infantile spasms, non-specific autistic features, and a milder phenotype involving some preserved language function and ambulation. In members of a family with the same mutation, there can be vast heterogeneity: in a case-series report, one affected girl had features of atypical RTT while her monozygotic twin sister only had autistic features, and their affected brother had a severe epileptic encephalopathy [4]. Many authors have commented that dysmorphic features are not consistently observed, while at least one series has tabulated a list of features, such broad forehead, high hairline, everted upper or lower lip, prominent nasal tip, long tapered fingers and more [17, 19, 21, 30, 31]. As CDKL5 is widely expressed in the body, there are other associated systemic features, similar to RTT. These may include gastrointestinal disturbances such as constipation and gastroesophageal reflux disease, autonomic dysfunction, breathing abnormalities, bruxism, dysphagia and impaired sleep [13, 17, 32, 33].
There has been a major effort in recent years to emphasize unique features in CDD compared to RTT and other similarly presenting syndromes. Seizure onset by 3-6 months of age is one of the first features of disease, in contrast to RTT which usually presents with developmental or behavioural features with seizures developing after 1 year of age if at all [20]. As others have noted, this is reminiscent of the old nomenclature of Hanefeld variant RTT, also formerly classified as early-seizure variant RTT [34]. In CDD, there is no period of normal development with late childhood attainment of milestones, if ever, in contrast to RTT that features normal development followed by a period of rapid regression and then plateauing [20, 35]. Males with CDD and RTT are infrequently reported. Males with CAEE can have phenotypes with the same severity as females, as opposed to RTT in males, which is more severe and usually fatal [17]. However, a recent report from an international registry found a trend towards lower attainment of developmental milestones in males compared to females with CAEE [35, 36].
The electrophysiologic profile in CDD can be characteristic. Initial EEG findings can be normal with early infantile seizures progressing to infantile spasms. EEG abnormalities can change with age, as opposed to most other metabolic or genetic epileptic encephalopathies in infancy that present with more stable and grossly abnormal EEG findings [21].
The seizure semiology can also be distinct and evolves with age. Bahi-Buisson et al. characterized three stages of epilepsy: Stage 1, early infantile epilepsy involving generalized tonic-clonic seizures of short duration multiple times daily, often responsive to anti-epileptic therapy; Stage 2, epileptic encephalopathy in late infancy/toddlers with refractory seizure treatment and sometime infantile spasms, which coincides with the developmental regression stage; and Stage 3, late multifocal or myoclonic epilepsy in children and adolescents with either complete recovery of epilepsy or severe refractory epilepsy resistant to all anti-epileptics and the ketogenic diet [21]. More recently and observed in half of the patients in our series, the hypermotor-tonic-spasms sequence is a highly specific seizure sequence that has been observed in some patients in late infancy/toddlerhood (Figure 1) [37, 38]. Identification of this sequence is at times subtle and requires involvement of an epileptologist. Once identified, it could prompt the appropriate diagnostic workup as it did for two of our patients.
II Genetics
Suspected pathogenic mutations have been grouped based on their effects [12, 35]. This includes whole gene deletions or truncating mutations in the N-terminus resulting in non-functional protein, missense mutations within the kinase domain that render it non-functional and truncating mutations near the C-terminus that result in a truncated protein with or without some preserved C-terminus. CDD is X-linked dominant with variable expressivity of moderate to severe phenotypes. No clear genotype-phenotype relationship has been demonstrated [12, 35].
Similarly, no relationship between X-inactivation patterns and disease severity has been established in CDD. Futhermore, a case report describes the phenotypic discrepancy between affected monozygotic twin girls who both had a high degree of X-inactivation skewing of the same chromosome [4]. Different X-inactivation skewing patterns in the brain might account for the different phenotypic severity, but to our knowledge this has never been demonstrated either in humans or animal models. Given that the vast majority of cases are sporadic with only rare recurrence in families, it appears that most mutations are de novo and disease recurrence in subsequent children appears to be the result of germline mosaicism [4].
Conclusion
In this article, we present a case series of four patients with CDD, one with the unusual presentation of reflex seizures and three with novel mutations not previously reported. CDKL5-Defeciency Disorder has gained recognition in the last decade as a distinct severe developmental disorder associated with drug-resistant epileptic encephalopathy and Rett-like features. Recent publications have classified CDD as an independent clinical entity instead of considering it as a subtype of RTT or atypical RTT. As a result, patient support websites have emerged internationally, for example, link1 and link2. Recently, a CDKL5 handbook was published by CDKL5 Canada [33].
The 2010 ILAE guidelines on genetic testing for epilepsies recommends testing for CDKL5 mutations in children with early onset spasms in the first year of life, which is predicted to have a 10-17% diagnostic detection rate [16]. Therefore, in a patient presenting with a phenotype consistent with CAEE, consideration should be given to sequencing CDKL5 alone or as part of a next generation sequencing panel [15, 17, 18]. Although there is no treatment to prevent the disease, anti-seizure drugs, vagal nerve stimulator and the ketogenic diet are mainstays with variable success, and other systemic features are treated symptomatically. It is very difficult to recommend specific agents to achieve seizure control given the complex and evolving nature of the epileptic encephalopathy and limited data of consistent long-term success. As CAEE increasingly gains recognition as a distinct clinical entity, more data will become available about the clinical features and pharmacologic management. Ultimately, since a large part of the pathophysiologic process affects postnatal neurodevelopment and early diagnosis is becoming more realistic, we are optimistic that active ongoing laboratory research has the potential to lead to targeted therapies.
Acknowledgements
Thank you to the families, EEG staff and colleagues for allowing us to participate in the care of their children and for their helpful discussions about the topic, allowing us to learn together.
Highlights
• A case series of four patients with CDKL5-associated epileptic encephalopathy (CAEE) or CDKL5 Deficiency Disorder (CDD) is presented. This includes one patient with reflex seizures as a rare manifestation of CAEE and three patients with mutations not previously reported.
• A recently recognized clinical seizure sequence specific to CAEE or CDD is described in detail.
• A review of clinical and molecular features is presented to aid the clinician in early recognition and diagnosis.
Funding
None.
Conflicts of Interest
None.
Article Info
Article Type
Case SeriesPublication history
Received: Mon 30, Mar 2020Accepted: Fri 17, Apr 2020
Published: Thu 30, Apr 2020
Copyright
© 2023 Juan Pablo Appendino. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.GG.2020.01.02
Figures & Tables
Table 1: Patients’ clinical features.
|
|
Patient 1 - “Reflex seizures” |
Patient 2 – “Novel frameshift” |
Patient 3 – “Novel splice site” |
Patient 4 – “Novel nonsense” |
|
Seizure onset |
6 months |
2 months |
2 months |
3 months |
|
Sex |
Female |
Female |
Female |
Female |
|
Family history |
Unremarkable |
Unremarkable |
Unremarkable |
Unremarkable |
|
Seizure semiology |
6 months-18 months: startle spasms sometimes progressing to GTC only with bathing 18 months: tonic-spasms sequence sometimes progressing to GTC spontaneous or startle to submersion in water and loud noise. No hypermotor component. |
2 months-2.5 years: Tonic, focal, autonomic symptoms 2.5 years: seizures in clusters |
2 months-10 months: focal-tonic 10 months: hypermotor-tonic-spasm sequence |
3 months-12 months: GTC 12 months-1.5 years: hypermotor-tonic-spasm sequence 1.5 years-2 years: tonic-spasm 2 years: GTC, focal |
|
EEG findings |
8 months: Excess high amplitude background slowing, somewhat poorly developed sleep features and parietal region hemispheric asymmetry 2 years: Multiple independent spike foci (MISF) and slow background activity for age |
Delta slowing of the background followed by generalized attenuation during seizures and multifocal interictal epileptiform abnormalities |
Bilateral independent frontal interictal discharges during sleep. During ictal event, there was high amplitude 2 Hz delta (hypermotor), followed by diffused attenuation (tonic) and spasms complex afterward. (refer to Figure 1.). |
Bilateral independent frontal interictal discharges during sleep. No EEG of ictal events was ever captured. |
|
Development |
Severe delayed gross motor, fine motor, language and social |
Severe delayed gross motor, fine motor, language and social |
Severe delayed gross motor, fine motor and language Mild delay social |
Severe delayed gross motor, fine motor, language and social |
|
Stereotypies |
Abnormal hand movements to midline |
Hyperoral Hand flapping |
Abnormal hand movements to midline |
Abnormal hand movements to midline Obsession with hands |
|
Systemic disease |
GERD |
None |
None |
None
|
|
Physical examination |
Normal growth parameters Axial hypotonia, hyperreflexia Poor tracking/eye contact |
Microcephaly (<2 SD) Axial hypotonia |
Microcephaly (<2 SD) Axial hypotonia Intermittent dysconjugate strabismus |
HC decreased growth velocity Axial hypotonia |
|
Investigations |
NBS, lactate, uric acid, ammonia, plasma and urine amino acids, urine organic acids, succinylpurines, Batten disease screen of CLN1, CLN2 and ultrastructural examination for membrane bound inclusions, acylcarnitine profile, alpha galactosidase, chitotriosidase, galactocerebrosidase, urine and plasma creatine and guanidinoacetate, TIEF, lysosomal enzyme activities, palmitate oxidation and ATP synthesis, CSF analysis (neurotransmitters, lactate, glucose, protein, amino acids), MRI-brain with spectroscopy, array-CGH, MECP2 sequencing, CDKL5 MLPA and array-CGH |
NBS, lactate, lipoprotein profile, plasma and urine amino acids, urine organic acids, acylcarnitine profile, total and free serum carnitine levels, plasma ammonia, total plasma homocysteine, serum CK, liver enzymes, urine alpha-AASA, creatine, biotinidase, VLCFA, Batten disease screen, CSF analysis (amino acids, lactate, glucose, protein, cell count, neurotransmitters), MRI-brain with spectroscopy, karyotype, gene sequencing (SCN1A, MECP2, CDKL5) |
NBS, plasma and urine amino acids, total and free carnitine, acylcarnitine, urine organic acids, CBC, electrolytes, Ca, Mg, phosphate, lipase, liver enzymes, albumin, lactate, , uric acid, ammonium, VLCFA, homocysteine, CK, lysosomal enzyme activity, Krabbe, CLN1, CLN2, plasma pipecolic acid, CSF analysis (protein, glucose, lactate, neurotransmitters, pyridoxal 5’ phosphate, 5-HIAA, HVA), Vit E, Vit B12, Vit B6, urine methylmalonic acid, cholesterol, MRI-brain with spectroscopy, array-CGH, gene sequencing (PNPO, CDKL5) |
NBS, plasma and urine amino acids, urine organic acids, lactate, total and free carnitine, TIEF, creatine, GAA, sialic acid, lipase, CBC, electrolytes, Ca, Mg, phosphate, lipoproteins, liver enzymes, albumin, lactate, uric acid, ammonia, LCFA, VLCFA, homocysteine, CK, MRI-brain with spectroscopy, array-CGH, gene sequencing (PNPO, MECP2, CDKL5) |
|
Molecular diagnosis |
c.162_99del261 |
c.2480_2486dupCAGATCT |
c.99+1G>A |
c.1238C>A |
|
Age at diagnosis |
2 years |
2 years |
<1 year |
2 years |
|
Antiepileptics |
Phenobarbital, Topirimate, Clobazam, Valproate |
Phenobarbital, Topirimate, Clobazam, Valproate, Keppra, Vitamin B6, Phenytoin, Lamotrigine, Nitrazepam, Oxcarbazepine, Mirtazipine, KCI, Levetiracetam, ketogenic diet |
Phenobarbital, Topirimate, Clobazam, Levetiracetam, Lamotrigine, Vit B6, Vit B12, folate, ketogenic diet |
Phenobarbital, Topirimate, Clobazam, Valproate, Levetiracetam, Oxcarbazepine, Gabapentin |
|
Other medications |
Ranitidine |
Carnitine |
None |
None |
References
- Záhorákova D, Langova M, Brožová K, Laštůvková J, Kalina Z et al. (2016) Novel CDKL5 Mutations in Czech Patients with Phenotypes of Atypical Rett Syndrome and Early-Onset Epileptic Encephalopathy. Folia Biol (Praha) 62: 67-74. [Crossref]
- Christianto A, Katayama S, Kameshita I, Inazu T (2016) A novel CDKL5 mutation in a Japanese patient with atypical Rett syndrome. Clin Chim Acta 459: 132-136. [Crossref]
- Epileptic Encephalopathy Early Infantile, 2; EIEE2. (2016) OMIM.
- Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL et al. (2004) Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 75: 1079-1093. [Crossref]
- Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffmann K, Moser B et al. (2004) Mutations in the X-Linked Cyclin-Dependent Kinase–Like 5 (CDKL5/STK9) Gene Are Associated with Severe Neurodevelopmental Retardation. Am J Hum Genet 75: 1149-1154. [Crossref]
- Scala E, Ariani F, Mari F, Caselli R, Pescucci C et al. (2005) CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet 42: 103-107. [Crossref]
- Evans JC, Archer HL, Colley JP, Ravn K, Nielsen JB et al. (2005) Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet 13: 1113-1120. [Crossref]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ et al. (2010) Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 68: 944-950. [Crossref]
- Huppke P, Held M, Laccone F, Hanefeld F (2003) The spectrum of phenotypes in females with Rett Syndrome. Brain Dev 25: 346-351. [Crossref]
- Lucariello M, Vidal E, Vidal S, Saez M, Roa L et al. (2016) Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Hum Genet 135: 1343-1354. [Crossref]
- Romaniello R, Saettini F, Panzeri E, Arrigoni F, Bassi MT et al. (2015) A de-novo STXBP1 gene mutation in a patient showing the Rett syndrome phenotype. Neuroreport 26: 254-257. [Crossref]
- Kilstrup-Nielsen C, Rusconi L, La Montanara P, Ciceri D, Bergo A et al. (2012) What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast 2012: 728267. [Crossref]
- Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E et al. (2005) CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet 14: 1935-1946. [Crossref]
- Liu JS (2011) Molecular genetics of neuronal migration disorders. Curr Neurol Neurosci Rep 11: 171-178. [Crossref]
- Nemos C, Lambert L, Giuliano F, Doray B, Roubertie A et al. (2009) Mutational spectrum of CDKL5 in early-onset encephalopathies: a study of a large collection of French patients and review of the literature. Clin Genet 76: 357-371. [Crossref]
- Ottman R, Hirose S, Jain S, Lerche H, Lopes-Cendes I et al. (2010) Genetic testing in the epilepsies--report of the ILAE Genetics Commission. Epilepsia 51: 655-670. [Crossref]
- Fehr S, Wilson M, Downs J, Williams S, Murgia A et al. (2013) The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet 21: 266-273. [Crossref]
- Intusoma U, Hayeeduereh F, Plong-On O, Sripo T, Vasiknanonte P et al. (2011) Mutation screening of the CDKL5 gene in cryptogenic infantile intractable epilepsy and review of clinical sensitivity. Eur J Paediatr Neurol 15: 432-438. [Crossref]
- Archer HL, Evans J, Edwards S, Colley J, Newbury-Ecob R et al. (2006) CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet 43: 729-734. [Crossref]
- Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N et al. (2008) Key clinical features to identify girls with CDKL5 mutations. Brain 131: 2647-2661. [Crossref]
- Bahi-Buisson N, Kaminska A, Boddaert N, Rio M, Afenjar A et al. (2008) The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia 49: 1027-1037. [Crossref]
- Roche Martínez A, Alonso Colmenero MI, Gomes Pereira A, Sanmartí Vilaplana FX, Armstrong Morón J et al. (2011) Reflex seizures in Rett syndrome. Epileptic Disord 13: 389-393. [Crossref]
- Solazzi R, Fiorini E, Parrini E, Darra F, Dalla Bernardina B et al. (2018) Diaper changing-induced reflex seizures in CDKL5-related epilepsy. Epileptic Disord 20: 428–433. [Crossref]
- Peikes T, Hartley JN, Mhanni AA, Greenberg CR, Appendino JP (2019) Reflex Seizures in a Patient with CDKL5 Deficiency Disorder. Can J Neurol Sci 46: 482–485. [Crossref]
- Russo S, Marchi M, Cogliati F, Bonati MT, Pintaudi M et al. (2009) Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 10: 241-250. [Crossref]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285-291. [Crossref]
- Lin C, Franco B, Rosner MR (2005) CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet 14: 3775-3786. [Crossref]
- Richards S, Aziz N, Bale S, Bick D, Das S et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405-424. [Crossref]
- Liang J-S, Shimojima K, Takayama R, Natsume J, Shichiji M et al. (2011) CDKL5 alterations lead to early epileptic encephalopathy in both genders. Epilepsia 52: 1835-1842. [Crossref]
- Mei D, Marini C, Novara F, Bernardina BD, Granata T et al. (2010) Xp22.3 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia 51: 647-654. [Crossref]
- Castrén M, Gaily E, Tengström C, Lähdetie J, Archer H et al. (2011) Epilepsy caused by CDKL5 mutations. Eur J Paediatr Neurol 15: 65-69. [Crossref]
- White R, Ho G, Schmidt S, Scheffer IE, Fischer A et al. (2010) Cylklin-dependent Kinase-Like 5 (CDKL5) Mutation Screening in Rett Syndrome and Related Disorders. Twin Res Hum Genet 13: 168-178. [Crossref]
- CDKL5 Canada. (2014) CDKL5 Disorder: A Guide for Parents, Canadian Edition.
- Hanefeld F (1985) The clinical pattern of the rett syndrome. Brain Dev 7: 320-325. [Crossref]
- Fehr S, Leonard H, Ho G, Williams S, de Klerk Net al. (2015) There is variability in the attainment of developmental milestones in the CDKL5 disorder. J Neurodev Disord 7: 2. [Crossref]
- IRSA. InterRett - IRSA Rett Phenotype Database.
- Melani F, Mei D, Pisano T, Savasta S, Franzoni E et al. (2011) CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol 53: 354-360. [Crossref]
- Klein K, Yendle SC, Harvey AS, Antony JH, Wallace G et al. (2011) A distinctive seizure type in patients with CDKL5 mutations: hypermotor-tonic-spasms sequence. Neurology 76: 1436-1438. [Crossref]