Almorexant, an Orexin Antagonist with Anti-Tumoral Properties in Human Colon Cancer
A B S T R A C T
Colorectal cancer, which is the third most common cancer, is the main cause of digestive cancer death. Previous studies have demonstrated that orexins, hypothalamic neuropeptides involved in sleep and food intake regulations, have anti-tumoral properties in digestive cancers. In the present work, we investigated the anti-tumoral role of an orexin antagonist, almorexant, in colon cancer. The anti-tumoral role of almorexant has been determined by in vitro and in vivo studies using HT-29 colon cancer cell line, which expressed endogenous orexin receptor 1 subtype (OX1R). Our in vitro study indicated that almorexant was able to reduce HT-29 cell viability by induction of mitochondrial apoptosis involving the tyrosine phosphatase SHP2 and the p38 signaling pathways. In contrast, no effect was observed in the colon cancer cell line HCT-116, which does not express OX1R, demonstrating that the anti-tumoral effect of almorexant was mediated by OX1R. When HT-29 cells were xenografted in nude mice, the administration of almorexant strongly reduced the tumor development with a potency similar to orexin. Our study supports that almorexant, a small molecule analog of orexin peptide, could represent a putative candidate in the treatment of colorectal cancer.
Keywords
Orexins, neuropeptides, GPCR, OX1R, colon cancer, apoptosis, biased ligand, pharmacology
Introduction
Colon cancer, also termed colorectal cancer (CRC), represents the third common cause of cancer across the world and displays the second cause of cancer death. The exact cause of CRC is unknown, but various factors such as age, personal or family history, obesity, the presence of colon polyps, tobacco or alcohol consumption, viral or bacterial infections, intestinal bowel disease, including Crohn’s disease or ulcerative colitis, and sedentary lifestyle were reported to increase the risk to develop such cancer [1]. Inversely, epidemiologic studies have shown, to varying degrees, the protective effect of many factors, including physical activity, diet rich in fruits and vegetables, vitamin supplements, coffee, garlic and drugs such as aspirin or non- steroidal anti-inflammatory drugs [2]. In early and advanced stages, patients with CRC undergo surgery followed by chemo- (using cytotoxic molecules as oxaliplatin, irinotecan) and/or radiotherapy in case of advanced stages [3].
Immunotherapy with recent antibodies targeting PD-1/PD-L1, in combination with the classical treatment, including antibodies targeting VEGF/VEGFR or EGF/EGFR pathways, represents promising strategies of treatment [4, 5]. Classically, the role of tumor-suppressor and oncogenes in which mutations and/or modified expression have been identified play a key role in the CRC development of [6]. Besides the involvement of these genes in the initiation and progression of CRC, roles of growth factors, hormones and signaling peptides, which interact with tyrosine kinase receptors or G protein- coupled receptors (GPCRs), also play a key role in tumor development [7]. GPCRs represent the largest family of membrane receptors encompassing about 800 members [8]. Some GPCRs are often over- or under-expressed and also ectopically expressed in tumors [7, 9-13]. The activation of these receptors by various signaling molecules, including among others gastrin, serine proteases, lipids, neurotensin, chemokines or prostaglandins lead to cancer cell growth through G protein transduction and/or transactivation of EGFR [10-17].
Among this large family of peptide hormones, orexins, also named hypocretins, which are hypothalamic neuropeptides, display anti-tumoral properties in CRC [18, 19]. The prepro-orexin precursor produces two isoforms termed orexin-A and orexin-B (hypocretin-1 and hypocretin-2), composed of 33 and 28 amino acids, respectively [20]. Orexins act mainly in the central nervous system, inducing the regulation of sleep/wakefulness state, reward seeking, the energy homeostasis and drug addiction [21, 22]. A dysregulation of the orexins system is involved in narcolepsy, a pathology of sleep [23].
Various studies have shown that orexins regulate in peripheral tissues various biological functions such as gastrointestinal motility, endocrine secretions, metabolism, blood pressure and reproductive functions [24]. Orexin-A (OxA) and orexin-B (OxB) interact with two GPCR sub-types termed OX1R and OX2R which are coupled to Gq signaling pathway leading to an increase of intracellular Ca2+level [25]. A few years ago, our group had demonstrated that OX1R was highly expressed in digestive cancers including, colon, pancreas, and liver cancers [19, 26, 27]. Activation of OX1R by orexins in cancer cell lines established from colon, pancreas and liver cancers led to mitochondrial apoptosis independently of the Ca2+ canonic signaling pathway [19]. This orexin- induced apoptosis was triggered by the phosphorylation of two ITIMs (immunoreceptor tyrosine-based inhibitory motifs) present in the OX1R sequence leading to the recruitment and the activation of the tyrosine phosphatase SHP2, and to the activation of p38 signaling pathway, which result in the formation of mitochondrial apoptosome leading to the activation of caspases-3 and -7 [28].
We have previously shown using a preclinical model that orexins display anti-tumoral properties when cancer cell lines derived from colon, pancreas, liver, and prostate cancers were sub-cutaneous xenografted in mice [27]. Over the past decade, orexin receptor targeting molecules, in particular, antagonists, have been developed to treat insomnia [28, 29]. Two classes of antagonists have been designed as single orexin-receptor antagonists (SORAs) and dual orexin-receptor antagonists (DORAs), which depending on the ability act on one or both orexin receptor subtypes [30]. Suvorexant, a DORA molecule, has recently obtained the approval of the U.S. Food & Drug Administration (FDA) for the treatment of insomnia [31]. This antagonist inhibits the intracellular Ca2+release, which represents the main signaling pathway involved in sleep regulation [32]. Surprisingly, another DORA molecule, named almorexant of the same class as suvorexant, was able to inhibit cellular growth by apoptosis induction in pancreas cancer, suggesting that this antagonist, but also suvorexant to a lesser degree, behave as biased ligands [26]. The present report shows that almorexant was able to induce apoptosis in the colon cancer cell lines HT-29 and also inhibited tumors development when they are xenografted in nude mice. These data indicate that almorexant could be regarded as a new therapeutic molecule in colon cancer and, more generally, in digestive cancers.
Materials and Methods
I Cell Line Culture and Treatments
The human colon cancer cell lines, HT-29 and HCT-116, were obtained from the American Type Culture Collection (ATCC, LGC Standards, Molsheim, France). HT-29 and HCT-116 cells were routinely grown in 25 cm2 plastic flasks (TPP, Techno Plastic Products, Trasadingen, Switzerland) containing Dulbecco Modified Eagle Medium (DMEM) which is deprived of pyruvate and supplemented with 4.5 g/L of glucose, 10 % (v/v) fetal calf serum, 100 µg/ml streptomycin, 100 units/ml of penicillin and 1% (v/v) ZellShield (Minerva Biolabs, Berlin, Germany) to prevent the mycoplasma contamination. Classically, HT-29 and HCT-116 cells were seeded in a 24-well plate at 50,000 cells per well and were maintained at 37°C in a humidified 5% CO2sub/air incubator. After 24/48 h of growth, cells were treated with 1 µM of OxA (GL Biochemicals, Shanghai, China) or 1 µM of almorexant or 1 µM suvorexant (MedChemExpress, Sollentuna, Sweden) for 48h and cell viability was determined by counting surviving cells.
II Caspase-3 Activity Detection
HT-29 cells were pretreated 24h with or without 50 µM pan- SHP inhibitor NSC87877 or 10 µM specific SHP2 inhibitor RMC4045 or were pretreated 30 min or not with 10 µM p38 inhibitor PD169316 and then, treated for 24h with or without 1 µM OxA or 1 µM almorexant. Caspase-3 activity detection was performed according to the manufacturer’s instructions using the caspase-3 assay colorimetric kit (#ab39401, Abcam, Paris, France).
III Preclinical Studies
2.106 HT-29 cells/100 µL PBS were inoculated subcutaneously into the flank of nude mice previously anesthetized by isoflurane 3%. Tumor development was followed by 2-dimensional caliper measurements (length (L) and width (W)). The tumor volume (V) was calculated by the formula of prolate ellipsoid as V=LW2π/6. PBS buffer (control mice) or 100 mg/kg of OxA or 100 mg/kg almorexant were intraperitoneally injected 3 times per week for 50 days. The bodyweight of mice after 50 days was similar in the untreated animal batch (44.9 ± 1.5 g, n=4) vs. the OxA treated animal batch (44.4 ± 0.7 g, n=4) and vs. almorexant treated animal batch (45.1 ± 1.2 g, n=4). Animal experiments were carried out according to the commonly accepted '3Rs' rule and complied strictly with the European legislation (APAFiS agreement N°#17199).
IV Statistical Analysis
The statistical analysis was determined by the Mann-Whitney non- parametric tests. The studies comprising more than two categories were analysed by ANOVA (analysis of variance) tests. All data were compiled and tested with the Prism 8.2.1 software (GraphPad Software, San Diego, CA, USA). The limit of statistical significance was set at p < 0.05.
Results and Discussion
I Almorexant and Suvorexant Inhibited the Cell Growth of HT-29 Colon Cancer Cell Line
To investigate the role of almorexant and suvorexant in colon cancer, the HT-29 colon cancer cell line was used. As previously demonstrated, OX1R and OxA were able to inhibit the growth of this cancer cell line [19]. Incubation of these cells with 1 µM almorexant or 1 µM suvorexant revealed that these antagonists were also able to inhibit HT-29 cell growth (Figure 1). It should be noted that almorexant and suvorexant induced 34 ± 4% and 20 ± 8% of HT-29 cell death, respectively (Figure 1). Moreover, almorexant activity was similar to OxA effect on cell death, i.e., 39 ± 6% (Figure 1). Based on these results indicating that almorexant was more efficient than suvorexant to inhibit the HT-29 cell growth, the next experiments were carried out using only almorexant. The inhibition of colon cancer cell growth induced by almorexant was dose-dependent (Figure 1).
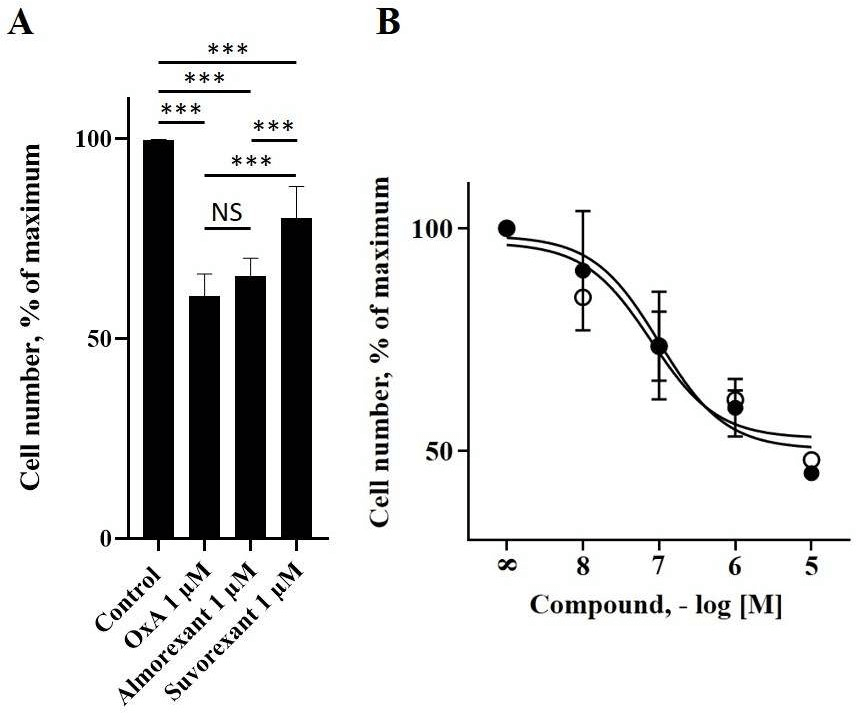
Figure 1: Almorexant, suvorexant and OxA inhibited the cell viability of the colon cancer cell line, HT-29. Panel A, 50,000 HT-29 cells were challenged or not (control) with 1 µM OxA,1 µM almorexant or 1 µM suvorexant for 48h. Panel B, dose response of OxA (○) or almorexant (●) on cell viability of HT-29 cells after 48h of treatment. Cell viability was determined by cell counting.
Results were expressed as percentage of maximal viability ± SE of three separate experiments.
***: p<0.001; NS: non-significant.
The determination of EC50 indicated that almorexant induced HT-29 cell death with an EC50 = 52 ± 20 nM, whereas OxA induced HT-29 cell death with an EC50 = 74 ± 27 nM, which were not significantly different. In contrast, when HCT-116 cells, a colonic cancer cell line which did not express OX1R, were incubated in the presence of almorexant or OxA, did not have any effect on cell growth as observed on (Figure 2) [19]. This absence of effect demonstrated that the inhibition of cell growth induced by almorexant in colon cancer cells is dependent on OX1R expression, as previously demonstrated for orexins in HT-29 cells [26]. These data indicated that almorexant-induced colon cancer cell growth inhibition was effectively mediated by OX1R.
II Almorexant Induced Mitochondrial Apoptosis in the Colon Cancer Cell Line HT-29
As shown in (Figure 2), the inhibition of HT-29 cell growth was associated with apoptosis induction by almorexant. Almorexant triggered apoptosis in HT-29 cells through increased caspase-3 activity similar to OxA (Figure 2). These effects induced by almorexant and OxA were totally abolished when HT-29 cells were pretreated by NSC87877, a pan-SHP inhibitor, or by RMC4045, a specific inhibitor of SHP2 (Figure 2). Moreover, the pretreatment of HT-29 cells with PD169316, a specific inhibitor of p38 that regulates the p38-MAPK signaling pathway, totally reverted the activation of caspase-3 induced by almorexant or OxA (Figure 2). Conventionally, GPCRs induce their cellular effects through a canonical signaling pathway involving classical second messengers such as intracellular Ca2+, or cAMP [33]. However, it is well-accepted that GPCRs can also mediate various biological effects via a non-canonical signaling pathway [34]. This notion of GPCR multi-coupling to various signaling pathways gave rise to the development of a new class of ligands able to distinguish these different signaling pathways [35].

Figure 2: OxA and almorexant induced cell apoptosis in HT-29 cell line but not in HCT- 116 cell line. Panel A, HCT-116 cells were grown in the absence (control) or in the presence of 1 µM OxA or 1 µM almorexant for 48h and then, cell viability was determined by cell counting. Results were expressed as percentage of maximal viability. Panel B, 300,000 HT-29 cells were pretreated or not (control) 24h with 50 µM NSC-87877 or 10 µM RMC4045 or 10µM PD169316 and then, treated or not (white bars) with 1 µM OxA (black bars) or 1 µM almorexant (grey bars). Caspase-3 was determined using a colorimetric assay kit (see Materials and methods).
Results are means ± SE of three separate experiments.
*: p<0.05; **: p<0.01; NS: non-significant.
These ligands, named “biased ligand”, could stabilize receptor conformations leading to the activation of only one signaling pathway independently of other signaling pathways activated by the same receptor [36]. Until today, the main development of orexin receptor-targeting molecules related to its action on sleep/wake regulation was oriented to the production of non- antagonists named SORA encompassing SORA1 which are specific of OX1R and SORA2 which are specific of OX2R. Moreover, DORA antagonists, which are specific to both receptor subtypes, have also been developed [30]. These molecules, including almorexant, inhibited the canonical signaling pathway leading to the intracellular Ca2+release [26]. Previous and present data indicated that almorexant and, to a lesser extent, suvorexant were able to induce cellular apoptosis in the pancreas and colon cancer cells similarly to orexins [26]. This induction of mitochondrial apoptosis was dependent on the phosphorylation of two ITIMs present in OX1R, the recruitment of the tyrosine phosphatase SHP2, the activation of p38 signaling pathway leading to the cytochrome c release from mitochondria and the activation of caspase-3 and -7 [37]. It should be noted that the phosphorylation of ITIMs by SRC kinases was dependent on the β/γ subunits of Gq protein [37].
Moreover, the use of SHP2 and p38 inhibitors totally reverse the pro-apoptotic effect of almorexant in colon cancer cells demonstrating that almorexant activated the pro-apoptotic signaling pathway induced by OX1R as previously described [19]. Structure-function relationship studies of orexin peptide have shown that some residues such as S18, N20 and T27 were able to discriminate the Ca2+signaling pathway of the pro-apoptotic signaling pathway associated with OX1R [38]. Taken together these observations, suggest that almorexant, through molecular determinants of OX1R, was able to block the activation of αq/phospholipase C (PLC) leading to intracellular Ca2+release, and to activate the pro-apoptotic signaling pathway through β/γ subunits conferring on this molecule the property of biased ligand.
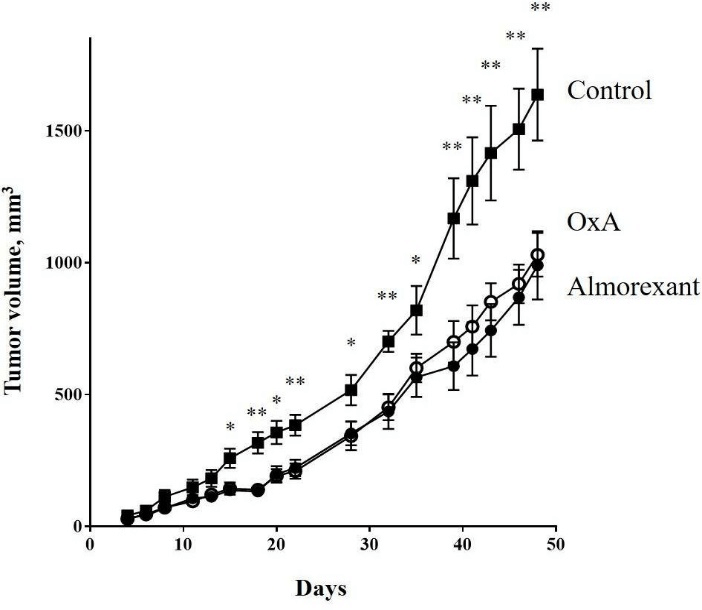
Figure 3: -29 cells (2.106) were subcutaneously inoculated into nude mice at day 0. Mice were intraperitoneally injected 3 times by week with 100 µl of PBS (■) or 100 µl of OxA solution (○) or 100 µl of almorexant solution (●). The OxA and almorexant treatments corresponded to 100 mg/kg. Data are means ± SE of 6 tumors in each group. For clarity, only statistical analysis between control, OxA and almorexant conditions was displayed. The statistical analysis between OxA and almorexant conditions indicated no significant differences.
*: p<0.05; **: p<0.01.
III Almorexant has Anti-Tumoral Properties in Colon Cancer Tumors Developed in Preclinical Models
To investigate the anti-tumoral role of almorexant, athymic nude mice were subcutaneously xenografted with the colon cancer cell line, HT-29 and then intraperitoneally injected 3 times/week with 100 mg/kg of almorexant or OxA. As shown in (Figure 3), almorexant was able to inhibit the tumor growth of about 40%. Moreover, almorexant had anti-tumoral effect very similar to OxA (Figure 3). These results indicated that almorexant, which was a full agonist of the pro-apoptotic signaling pathway activated by OX1R, had an equal potency than OxA to induced cellular apoptosis and anti-tumoral action in colon cancer development. However, almorexant had various advantages compare to the native peptide. Indeed, OxA was very sensitive to degradation; its half-life time was estimated as 27 min in serum, whereas almorexant had an estimated half-life time of about 32 h, which is a crucial advantage [39, 40]. OxA is a full agonist and induced a full activation of canonical and non-canonical signaling pathways. These activations led to the various biological actions of orexins in the central nervous system and in peripheral tissues where orexins have some actions on cardiovascular, genitourinary and digestive tracts [24]. A putative obstacle that reduces the therapeutic use of orexins in cancer treatment will be the existence of side effects having deleterious impacts.
However, a recent in vivo study using mice showed that orexin-A treatment could reduce fat mass but does not any significantly affect the components of energy expenditure [41]. Moreover, some reports indicated that orexins could activate other signaling pathways involving MAPK- Erk1/2, cAMP, PI3K-Akt and JNK [42]. In contrast, almorexant which activated mainly the pro-apoptotic signaling pathway but blocked the PLC signaling pathway, could limit the side effects which could be induced by intracellular Ca2+production. Taken together, these observations suggest that orexin receptor antagonists could be a new innovative approach to treat digestive cancers and, in particular, colon cancer.
Conclusion
We have demonstrated that the orexin antagonist almorexant was able to activate the pro- apoptotic signaling pathway involving the recruitment of SHP2 in colon cancer cell line, HT-29. Moreover, in the preclinical model, almorexant was able to induce an anti-tumoral effect comparable to orexins properties as previously described [19]. In conclusion, almorexant, a biased ligand, could represent an innovative therapeutic approach in colon cancer treatment.
Acknowledgements
The study on almorexant and orexins in digestive cancers was supported in part by Inserm Transfert, INSERM U1149/ The Inflammation Research Center (CRI), The “Institut National du Cancer (INCA)” [grant number N°2013-213] and the “Ligue Nationale Contre le Cancer” [grant numbers R16020HH, GB/MA/CD/EP-12062].
Conflicts of Interest
None.
Article Info
Article Type
Research ArticlePublication history
Received: Thu 23, Apr 2020Accepted: Mon 08, Jun 2020
Published: Fri 24, Jul 2020
Copyright
© 2023 Alain Couvineau. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.EJMC.2020.01.02
Figures & Tables
Results were expressed as percentage of maximal viability ± SE of three separate experiments.
***: p<0.001; NS: non-significant.
Results are means ± SE of three separate experiments.
*: p<0.05; **: p<0.01; NS: non-significant.
*: p<0.05; **: p<0.01.
References
- Bostman FT (2014) Colorectal cancer (Chapter 5.5). In World Cancer Report, Stewart, B.W. and Wild C.P. Eds, Int Agency Res Cancer 392-402.
- Hadjipetrou A, Anyfantakis D, Galanakis CG, Kastanakis M, Kastanakis S (2017) Colorectal cancer, screening and primary care: A mini literature review. World J Gastroenterol 14: 6049-6058. [Crossref]
- Siegel R, Desantis C, Jemal A (2014) Colorectal cancer statistics, 2014. CA Cancer J Clin 64: 104-117. [Crossref]
- Yaghoubi N, Soltani A, Ghazvini K, Hassanian SM, Hashemy SI (2019) PD-1/ PD-L1 blockade as a novel treatment for colorectal cancer. Biomed Pharmacother 110: 312-318. [Crossref]
- Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausová J et al. (2012) Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 30: 3499-3506. [Crossref]
- Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361: 2449-2460. [Crossref]
- Insel PA, Sriram K, Wiley SZ, Wilderman A, Katakia T et al. (2018) GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Front Pharmacol 9: 431. [Crossref]
- Couvineau A, Laburthe M (2012) VPAC Receptors: Structure, Molecular pharmacology and interaction with accessory proteins. Br J Pharmacol 166: 42-50. [Crossref]
- Laburthe M, Rousset M, Boissard C, Chevalier G, Zweibaum A et al. (1978) Vasoactive Intestinal Peptide: A potent stimulator of adenosine 3’:5’-cyclic monophosphate accumulation in gut carcinoma cell lines in culture. Proc Natl Acad Sci U S A 75: 2772-2775. [Crossref]
- Maoret JJ, Pospaï D, Rouyer Fessard C, Couvineau A, Laboisse C et al. (1994) Neurotensin receptor and its mRNA are expressed in many human colon cancer cell lines but not in normal colonic epithelium: binding studies and RT-PCR experiments. Biochem Biophys Res Commun 203: 465-471. [Crossref]
- Singh P, Dai B, Wu H, Owlia A (2000) Role of autocrine and endocrine gastrin-like peptides in colonic carcinogenesis. Curr Opin Gastroenterol 16: 68-77. [Crossref]
- Darmoul D, Gratio V, Devaud H, Lehy T, Laburthe M (2003) Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am J Pathol 162: 1503-1513. [Crossref]
- Gratio V, Walker F, Lehy T, Laburthe M, Darmoul D (2009) Aberrant expression of proteinase-activated receptor 4 promotes colon cancer cell proliferation through a persistent signaling that involves Src and ErbB-2 kinase. Int J Cancer 124: 1517-1525. [Crossref]
- Yang M, Zhong WW, Srivastava N, Slavin A, Yang J et al. (2005) G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the{beta}-catenin pathway. Proc Natl Acad Sci U S A 102: 6027-6032. [Crossref]
- Sarvaiya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS (2013) Chemokines in tumor progression and metastasis. Oncotarget 4: 2171-2185. [Crossref]
- Karpisheh V, Nikkhoo A, Hojjat Farsangi M, Namdar A, Azizi G et al. (2019) Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat 144: 106338. [Crossref]
- Lappano R, Maggiolini M (2011) G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov 10: 47-60. [Crossref]
- Rouet Benzineb P, Rouyer Fessard C, Jarry A, Avondo V, Pouzet C et al. (2004) Orexins acting at native OX(1) receptor in colon cancer and neuroblastoma cells or at recombinant OX(1) receptor suppress cell growth by inducing apoptosis. J Biol Chem 279: 45875-45886. [Crossref]
- Voisin T, El Firar A, Fasseu M, Rouyer Fessart C, Descatoire V et al. (2011) Aberrant expression of OX1 receptors for orexins in colon cancers and liver metastases: an openable gate to apoptosis. Cancer Res 71: 3341-3351. [Crossref]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM et al. (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and g protein-coupled receptors that regulate feeding behavior. Cell 92: 573-585. [Crossref]
- de Lecea L, Sutcliffe JG (1999) The hypocretins/orexins: novel hypothalamic neuropeptides involved in different physiological systems. Cell Mol Life Sci 56: 473- 480. [Crossref]
- Mieda M, Yanagisawa M (2002) Sleep, feeding, and neuropeptides: roles of orexins and orexin receptors. Curr Opin Neurobiol 12: 339-345. [Crossref]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T et al. (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98: 437-451. [Crossref]
- Heinonen MV, Purhonen AK, Mäkelä KA, Herzig KH (2008) Functions of orexins in peripheral tissues. Acta Physiol (Oxf) 192: 471-485. [Crossref]
- Voisin T, Rouet Benzineb P, Reuter N, Laburthe M (2003) Orexins and their receptors: structural aspects and role in peripheral tissues. Cell Mol Life Sci 60: 72-87. [Crossref]
- Dayot S, Speisky D, Couvelard A, Bourgoin P, Gratio V et al. (2018) In Vitro, in vivo and ex vivo demonstration of the antitumoral role of hypocretin-1/orexin-A and almorexant in pancreatic ductal adenocarcinoma. Oncotarget 9: 6952-6967. [Crossref]
- Couvineau A, Dayot S, Nicole P, Gratio V, Rebours V et al. (2018) The anti-tumoral properties of orexin/hypocretin hypothalamic neuropeptides: an unexpected therapeutic role. Front Endocrinol (Lausanne) 9: 573. [Crossref]
- Couvineau A, Voisin T, Nicole P, Gratio V, Abad C et al. (2019) Orexins as novel therapeutic targets in inflammatory and neurodegenerative diseases. Front Endocrinol (Lausanne) 10: 709. [Crossref]
- Kukkonen JP (2017) Orexin/hypocretin signaling. Curr Top Behav Neurosci 33: 17-50. [Crossref]
- Khoo SY, Brown RM (2014) Orexin/hypocretin based pharmacotherapies for the treatment of addiction: DORA or SORA? CNS Drugs 28: 713-730. [Crossref]
- Cox CD, Breslin MJ, Whitman DB, Schreier JD, McGaughey GB et al. (2010) Discovery of the dual orexin receptor antagonist [(7R)-4-(5-Chloro-1,3-Benzoxazol-2- Yl)-7-Methyl-1,4-Diazepan-1-Yl][5-Methyl-2-(2H-1,2,3-Triazol-2- Yl)Phenyl]Methanone (MK-4305) for the treatment of insomnia. J Med Chem 53: 5320-5332. [Crossref]
- Heidmann B, Gatfield J, Roch C, Treiber A, Tortoioli S et al. (2016) Discovery of highly potent dual orexin receptor antagonists via a scaffold-hopping approach. ChemMedChem 11: 2132-2146. [Crossref]
- Hilger D, Masureel M, Kobilka BK (2018) Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 25: 4-12. [Crossref]
- Pavlos NJ, Friedman PA (2017) GPCR Signaling and trafficking: the long and short of it. Trends Endocrinol Metab 28: 213-226. [Crossref]
- Bermudez M, Nguyen TN, Omieczynski C, Wolber G (2019) Strategies for the discovery of biased GPCR ligands. Drug Discov Today 24: 1031-1037. [Crossref]
- Wootten D, Christopoulos A, Marti Solano M, Babu MM, Sexton PM (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 19: 638-653. [Crossref]
- El Firar A, Voisin T, Rouyer Fessard C, Ostuni MA, Couvineau A et al. (2009) Discovery of a functional immunoreceptor tyrosine-based switch motif in a 7-transmembrane-spanning receptor: role in the orexin receptor OX1R-driven apoptosis. FASEB J 23: 4069-4080. [Crossref]
- Nicole P, Couvineau P, Jamin N, Voisin T, Couvineau A et al. (2015) Crucial role of the orexin-B C-terminus in the induction of OX1 receptor-mediated apoptosis: analysis by alanine scanning, molecular modelling and site-directed mutagenesis. Br J Pharmacol 172: 5211-5223. [Crossref]
- Ehrström M, Näslund E, Levin F, Kaur R, Kirchgessner AL et al. (2004) Pharmacokinetic profile of orexin A and effects on plasma insulin and glucagon in the rat. Regul Pept 119: 209-212. [Crossref]
- Hoever P, Hay J, Rad M, Cavallaro M, van Gerven JM et al. (2013) Tolerability, pharmacokinetics, and pharmacodynamics of single-dose almorexant, an orexin receptor antagonist, in healthy elderly subjects. J Clin Psychopharmacol 33: 363-370. [Crossref]
- Blais A, Drouin G, Chaumontet C, Voisin T, Couvelard A et al. (2017) Impact of Orexin-A Treatment on Food Intake, Energy Metabolism and Body Weight in Mice. PLoS One 12: e0169908. [Crossref]
- Kukkonen JP, Leonard CS (2014) Orexin/hypocretin receptor signalling cascades. Br J Pharmacol 171: 314-331. [Crossref]