Journals
Effects of lithium treatment on oxidative stress markers in mitochondrial complex I and complex III inhibition and after CO2 exposure in SH-SY5Y cells
A B S T R A C T
This study aims to evaluate the effects of lithium chloride (LiCl) on resistance to oxidative stress, cell proliferation, and COX-2 expression, both in the absence of carbon dioxide (CO2), rotenone and antimycin A and after exposure to them. SH-SY5Y cells were treated for 72 hours with LiCl (0.5 - 5 mM). Superoxide anion (O2-) and intracellular reactive oxygen species (ROS) production were measured via fluorescence. All the experiments were carried out under standard conditions, after 4 h of CO2 exposure or after 24 hours of incubation with rotenone and antimycin A: inhibitors of mitochondrial complex I and III respectively. At very low concentrations, LiCl decreased O2- production under normoxic conditions and after exposure to either CO2 or rotenone. LiCl was not observed to produce any changes in intracellular ROS levels under standard conditions or after either the rotenone or antimycin A treatment; but changes were observed after CO2 exposure. In addition, LiCl increased cell proliferation and modified expression of COX-2, but only under oxidative stress situations. In conclusion, the antioxidant effect of lithium might be due, at least in part, to its inhibition of O2- from complex I. Also, lithium decreases the vulnerability of SH-SY5Y cells to cell injury.
K E Y W O R D S
Lithium,SH-SY5Y,COX-2,Oxidative stress,Rotenone,CO2,Antimycin A
I N T R O D U C T I O N
Bipolar disorder is a life-threatening psychiatric illness characterized by mood disturbances with recurrent periods of mania, hypomania and depression. Although lithium has been used for over 60 years to treat this illness, the basis of its therapeutic effect remains unclear [1]. It has been suggested that lithium is neuroprotective, as it increases expression of the gene encoding the anti-apoptotic protein Bcl-2 and decreases expression of the genes coding for the pro-apoptotic proteins p53 and Bax, in both cerebellar granule cells and SH-SY5Y cells [2]. It has also been observed that therapeutically relevant concentrations of lithium inhibit cyclic AMP accumulation, decrease the production of amyloid-(Aβ) peptides, inhibit Aβ-induced stress and also both prevent Tau phosphorylation and exert neuroprotective effects by increasing resistance to oxidative damage [3-6]. Indeed, it has been shown that lithium stimulates progenitor proliferation in cultured brain neurons at low concentrations and could have therapeutic effect on acute and chronic brain injuries [7].
Oxidative stress, which occurs when the oxidant activity in a tissue overwhelms its endogenous anti-oxidation and protection mechanisms, is a major factor in the pathogenesis of different acute and chronic neurological diseases. When the capacity of the antioxidant systems of the cell, is exceeded, increased production of reactive oxygen species (ROS) triggers oxidative stress and can lead to cell death [8, 9].
A mediator of oxidative stress is the cellular superoxide anion (O2-) which can influence both physiological and pathological processes [10]. The cytotoxic mechanisms by which ROS induce neuronal damage may involve direct oxidative attack on cell macromolecules (proteins, lipids, DNA and sugars) and initiation or propagation of free-radical chain reactions [11]. It has also been suggested that mitochondrial dysfunction is involved in neurodegenerative disorders including Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis. In addition, different studies report altered mitochondrial function in schizophrenia and major depressive disorder [12]. There is accumulating evidence indicating increased mitochondrial respiration in bipolar mania which contrasts with decreased mitochondrial function in patients in the euthymic or depressive phase of illness [13]. Dysfunction of the mitochondrial electron transport chain (ETC) is generally associated with increased mitochondrial ROS production. Mitochondrial complex I and complex III are generally considered the most important contributors to mitochondrial ROS production in intact cells [14]. The complex I inhibitor rotenone has been shown to stimulate O2- production. Moreover, it appears that complex I can also directly catalyze H2O2 formation, whereas inhibition of complex III with antimycin A releases large amounts of O2-. However, complex I appears to constitute the main source of mitochondrial O2- under physiological conditions [14, 15]. It has been reported that lithium administration increases superoxide dismutase and catalase in various tissues, including several brain regions [16]. Also, therapeutically relevant lithium concentrations could favor neurogenesis and decrease the vulnerability of neuronal cells to cell injury by increasing their antioxidant defenses [6].
Several papers provide strong evidence for the hypothesis that the arachidonic acid (AA) cascade is a major target for drugs that are effective in bipolar disorder, particularly in the manic phase of the disease; but that targeting could be indirect [17]. AA is a lipid released from membrane phospholipids via both receptor-G protein-initiated activation of phospholipase A2 (PLA2) and cyclooxygenase (COX)-mediated production of the eicosanoid metabolites: prostaglandins and thromboxanes [18]. Two COX isoforms have been described: COX-1 is constitutively expressed and is thought to produce eicosanoids for normal physiological function; whereas COX-2 is induced in pathological conditions, often in response to pro-inflammatory agents [19]. Direct targeting of COX-2 activity and prostaglandin E2 formation has been put forward as a means of treating bipolar disorder [20]. COX-2 is the predominant isoform in the brain and spinal cord, where it is involved in synaptic signaling as well as cerebral blood flow and behavior and also in oxidative neuronal damage [1, 21-23].
CO2 is an important gaseous molecule which plays a cell signaling role in all organisms [24]. In vitro CO2 exposure can induce redox status alteration, which results from an increase in intracellular ROS generation [25]. The direct effects of oxidative stress associated with CO2 insufflation in neuroblastoma cells are as yet unexplored, although Montalto et al. showed that oxidative stress induced by CO2 exposure has a toxic effect on neuroblastoma cells, leading to DNA damage [26]. Thus, since SH-SY5Y cells, derived from human neuroblastoma, have been widely used as an in vitro model for the study of the protective effects of anti-apoptotic substances and neurodegenerative disorders they were selected here to study the neuroprotective effects of lithium [27, 28]. The aims of this study were to analyze the effects of therapeutically -relevant concentrations of lithium chloride (LiCl) on SHSY-5Y cell proliferation, on COX-2 expression and on inhibition to oxidative damage. In this sense, we study the role played by CO2 induced oxidative stress in neuroblastoma cells treated with LiCl. Since some dysfunctions of the mitochondrial ETC are associated with increased mitochondrial ROS production, we determined whether inhibition of mitochondrial complex I and complex III were affected by LiCl.
Materials and methods
SH-SY5Y Cell cultures
SH-SY5Y human neuroblastoma cells were purchased from American Type Cell Culture (ATCC, Manassas, VA, USA) and were cultured in MEM supplemented with 10% FBS, 2 mM L-glutamine, 500 μM sodium pyruvate, 50 units/ml penicillin and 50 μg/ml streptomycin. The cells were grown in a humidified incubator with 5% CO2 at 37ºC. The culture medium was changed every 2-3 days.
Hypoxic/reoxygenation experiments
SH-SY5Y cells were placed in a modified desiccating chamber connected to a continuous CO2 flow at a pressure of 15 mmHg (100%) for 4 hours (hypoxia). The chamber was located in an incubator set at 37ºC. After CO2 exposure, the cells were immediately transferred to a standard cell incubator with a normoxic atmosphere (reoxygenation) of 95% air and 5% CO2 for a further 24-hour incubation period. Non- treated cultures were maintained in parallel for the same time in a standard cell incubator (95% air/5% CO2) at 37 ºC to represent standard conditions [26].
Superoxide anion (O2-) Quantitation by hydroethidine assay
Hydroethidine (HE) (Life Technologies), a sodium borohydride-reduced derivative of ethidium bromide, was used to detect O2-, which converts it to ethidium bromide causing an increase in red fluorescence. The increase in fluorescence was measured on a Wallac Victor 2 1420 Multilabel Counter (PerkinElmer), using an excitation wavelength of 510 nm and an emission wavelength of 590 nm. Neuroblastoma cells were seeded onto 6-well plates (Greiner Bio-one), allowed to adhere for at least 24 h, and then incubated in Hanks’ balanced salt solution (HBSS) containing 5 μM HE, for 30 min at 37ºC in the dark to allow the dye to become loaded into the cells [29]. Subsequently, the cells were treated with phenol-free RPMI containing 0, 0.5, 1 or 5 mM LiCl. The cells reacted on addition of the LiCl and we allowed the reaction to proceed under incubation at 37ºC for 72 h. A cell sample without HE was incubated as a negative control. The generation of O2- was measured and reported as the relative fluorescence intensity.
Measurement of intracellular ROS generation by DCF-DA assay
Conversion of non-fluorescent chloromethyl-DCF-DA (2’,7’-dichlorofluorescein diacetate) (Sigma-Aldrich) to fluorescent DCF was used to monitor intracellular H2O2 production and other oxidants. DCF were quantified using a microplate reader (Wallac Victor 2 1420 Multilabel Counter (PerkinElmer), using an excitation wavelength of 480 nm and an emission wavelength of 535 nm. After 72 hours LiCl treatment, neuroblastoma cells in 6-well plates were incubated with 10 μM for 30 min at 37ºC. After two washes with PBS, the cells were harvested using non-enzymatic cell dissociation solution and resuspended in 500 μl of PBS supplemented with 0.1 M KH2PO4 and 0.5% Triton X-100. Cell debris was pelleted by centrifugation at 2000g for 10 min, and the supernatants were analyzed under fluorescein optics, since the fluorescence intensity is proportional to the amount of intracellular peroxide produced by the cells [29].
Treatment with rotenone and antimycin A
Concentrations of rotenone and antimycin A were selected based on both data from previous studies carried out in our department, and those obtained by other authors [30, 31].
After 72 hours of LiCl (0.5, 1 or 5 mM) treatment, the neuroblastoma cells were incubated for 24 h at 37°C with 5 M rotenone or 10 nM antimycin A. We then continued with the method used in the HE and DCF assays.
Western blotting
After LiCl treatment, to obtain whole cell extracts, the cells were lysed using cell lysis buffer (Cell Signaling) + PMSF. Immediately, the cells were scraped off the plate and the extract were transferred to Eppendorf tubes on ice. The samples were sonicated for 15 seconds, and centrifuged at 9,000 g at 4ºC for 10 min. The protein concentration was evaluated in the supernatant by the Lowry’ method and 10 μg of proteins was incubated with 0.3 M Tris-HCl, 5% SDS, 50% Glycerol and 100mM DTT at 96ºC for 5 min [32]. 10 μl of the sample were loaded on a 4%-12% polyacrylamide gel for 100 min at 140V and 400 mA, and transferred to nitrocellulose membranes. Later, the membranes were blocked with 5% nonfat dry milk in Tris-buffered saline, 0.1% Tween 20 (TBS/T), for an hour and then washed three times for 10 min with TBS/T. The membranes were incubated overnight at 4ºC with the primary monoclonal antibody with TBS/T - 5% BSA. The membranes were washed three times for 10 min with TBS/T and incubated for 60 min at room temperature, in continuous agitation with the secondary antibody, at the appropriate dilution in TBS/T; and washed again 3 times for 10 min with TBS/T.
Protein strips were incubated for 5 min with ECL Clarity from BioRad and analyzed using a LAS-3000 Imaging System (Fujifilm). The bands were quantified by densitometry analysis.
Measurement of cell proliferation
Cell growth was determined using the MTT assay [33]. Briefly, the tetrazolium salt MTT was added to the SH-SY5Y cells (1mg/ml final concentration). After 2 hours of incubation at 37ºC, the lysing buffer was added (20% SDS in 50% N,N-dimethyl formamide, pH: 4,7). After further overnight incubation at 37ºC, the optical densities were measured at 570 nm. The percentage survival was defined as [Abs (experimental–blank/Abs (control–blank)]x100 where “blank” was the value taken from wells without cells.
Statistical analysis
Data were expressed as mean ± S.E. Statistical significance was established by one-way analysis of variance (ANOVA), with the Tukey post hoc test comparing the means. When appropriate, Student’s t-test was employed to compare two groups. Differences with p < 0.05 were considered statistically significant
Results
Incubation time period and LiCl concentrations assayed
To discriminate between adverse and beneficial effects, LiCl concentrations ranging from 0.5 to 5 mM were used [6, 7].
As a preliminary experiment, we were interested in defining the optimum incubation time for LiCl. For this, we determined the production of O2- in SH-SY5Y cells using, 5 mM LiCl, for 24, 48, 72 and 96 h. In all the time periods we included a control to rule out problems of growth or cell death. Our results show that the maximum O2- production was observed after 72 h of incubation. So, we selected this incubation time in all the subsequent experiments (results not shown).
O2- generation by the HE assays
O2- generation in LiCl-treated neuroblastoma cells was monitored by the HE dyes assay. Our results showed a significant decrease of O2- production after 72 h of incubation with LiCl. They also showed a significant decrease significantly with all concentrations of lithium; it should be noted that even very low concentrations of LiCl (0.5 and 1 mM) inhibited O2- production significantly (Figure 1).
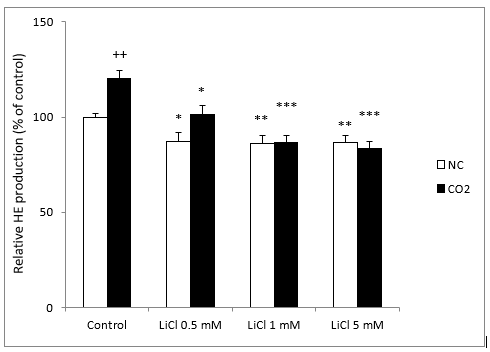
Figure 1: Fluorescence signal of HE oxidation products in SH-SY5Y cells in the presence or absence of CO2, following exposure for 72 hours to various concentrations of LiCl. Data are expressed as means ± S.E.M of six independent experiments performed in triplicate. *P < 0.05, **P < 0.01 and ***P < 0.001 compared LiCl results to their respective control group. ++P < 0.01 comparison of CO2 group versus normoxia conditions (NC) group.
Since CO2 exposure induced an increase in intracellular ROS generation in SH-SY5Y cells, which may cause oxidative damage, we tested the effects of different concentrations of LiCl, 24 hours after the cells were exposed to 15 mmHg CO2 for 4 hours. In this case, we observed a significant increase in O2- production in comparison with standard conditions. As in the standard group, all concentrations of lithium significantly decreased the production of O2- (Figure 1).
In the presence of rotenone (5 M), LiCl also inhibited O2- production in comparison to rotenone alone; showing a significant effect at concentrations of 0.5 mM of LiCl (Figure 2).
In contrast, in the presence of antimycin A (10 nM), LiCl did not modify O2- production at any of the LiCl concentrations assayed (Figure 2).
It should be noted that incubation with either rotenone or antimycin A both stimulated O2- production very significantly (Figure 2).
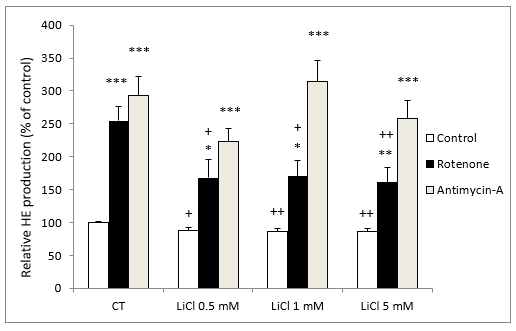
Figure 2: Fluorescence signal of HE oxidation products in SH-SY5Y cells following exposure for 72 hours to various concentrations of LiCl and subsequent treatment for 24 hours with rotenone (5 M) or antimycin A (10 nM). Data are expressed as means ± S.E.M of six independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001 comparing antimycin A and rotenone results to control group. + P < 0.05, ++ P < 0.01 significant differences compared with their respective control conditions group (CT).
Intracellular ROS generation by DCF assay
Intracellular production of ROS in lithium-treated SH-SY5Y cells was analyzed using the fluorescein derivative DCF dye assay for 72 hours. (Figure 3) shows ROS generation by neuroblastoma cells treated with LiCl. No statistically significant differences were found between any of the concentrations assayed.
When we exposed the cells to CO2, we observed a significant increase in ROS production in the control cells in comparison to normoxic conditions; whereas lithium, even at very low concentrations, significantly reduced this increase in ROS production. In this sense, LiCl decreased the effects of CO2 exposure very fast and LiCl 5 mM inhibited them completely (Figure 3).
In the same way as in the previous experiment, when we analyzed DCF production, no statistically significant differences were found between any of the concentrations of LiCl assayed. Neither were significant differences observed with lithium in the presence of rotenone (Figure 4). In the presence of antimycin A, a significant increase in ROS generation was only observed under the control conditions and for the 0.5 mM lithium: not with concentrations above this (Figure 4).
Measurement of cell proliferation by MTT
The results for cell proliferation show that LiCl significantly protected against cell death induced by CO2 exposure. The protection was dose-dependent and already statistically significant with 0.5 mM lithium (Figure 5). In contrast, in the normoxic group, a slight but significant difference was observed from 1 to 5 mM lithium (Figure 5). When we compared normoxic and CO2-treated cells, in all cases the differences were significant, although the results matched each other as we increased the LiCl concentration.
In the cells treated with rotenone, there was a tendency for lithium gradually to increase cell proliferation at all concentrations, but this was only significant at 1 and 5 mM (Figure 5). Something similar happened with antimycin A, as differences were observed from 1 mM LiCl (Figure 5). In the control groups, all the treatments assayed produced a significant decrease in cell proliferation in comparison with normoxic conditions. It is important to note that in all cases, LiCl gradually enhanced cell proliferation as the concentrations of this ion increased (Figure 5).
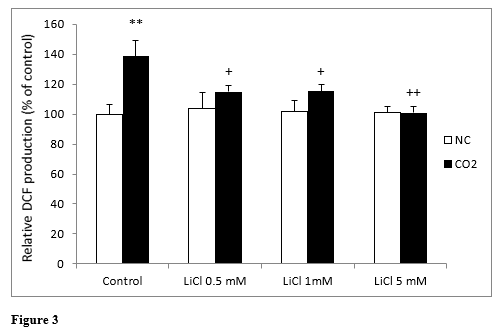
Figure 3: Changes in intracellular fluorescence signal of DCF oxidation products in SH-SY5Y cells in the presence or in the absence of CO2, following exposure for 72 hours to different concentrations of LiCl. Data are presented as the means ± S.E.M of six independent experiments performed in triplicate. **P < 0.01 comparing CO2 control group versus normoxia conditions (NC) group. +P < 0.05 and ++P < 0.01 significant differences compared LiCl with their respective control group.
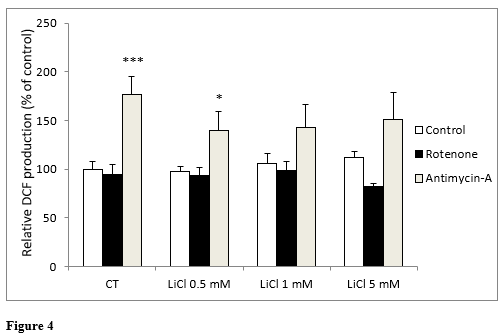
Figure 4: Changes in intracellular fluorescence signal of DCF oxidation products in SH-SY5Y cells following exposure for 72 hours to various concentrations of LiCl and subsequent treatment for 24 hours with rotenone (5 M) or antimycin A (10 nM). Data are presented as the means ± S.E.M of six independent experiments performed in triplicate. * P < 0.05, *** P < 0.001 comparing antimycin A and rotenone results to their respective control group.
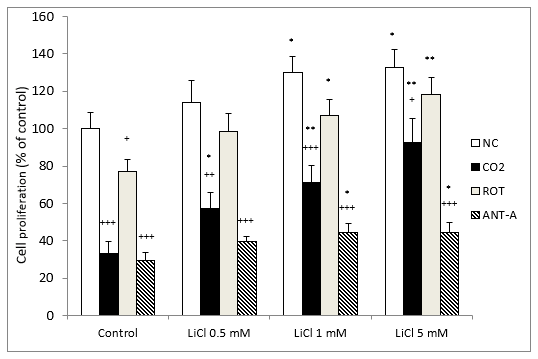
Figure 5: Dose-related effects of 72 hours exposure to LiCl concentrations ranging from 0.5 to 5 mM on SH-SY5Y cells’ growth rate in the absence or presence of CO2 and after subsequent treatment for 24 hours with rotenone (ROT) (5 M) or antimycin A (ANT-A) (10 nM). The cell viability corresponds to the mean ± S.E.M of six independent experiments performed in triplicate. + P < 0.05; ++ P < 0.01 and +++ P < 0.001 comparing CO2, rotenone and antimycin A groups versus normoxia conditions (NC) group. * P < 0.05, **P < 0.01, compared LiCl groups to their respective control group.
Comparison between NaCl and LiCl
To verify that the neuroprotective effect of lithium was specific and not due to other non-specific factors, we treated the cell cultures with sodium chloride (NaCl) and LiCl, both at 1 mM. In contrast to what happened with lithium, the sodium group showed no significant differences in intracellular ROS and O2- levels from the normoxia and CO2 exposure groups (results not shown).
When we compared the cell growth with LiCl and NaCl to the normoxia groups, we observed a slight but significant difference with the lithium treatment, but not with sodium. Meanwhile, in the CO2 group, LiCl produced a very significant increase in cell proliferation, while NaCl caused no significant differences (Figure 6).
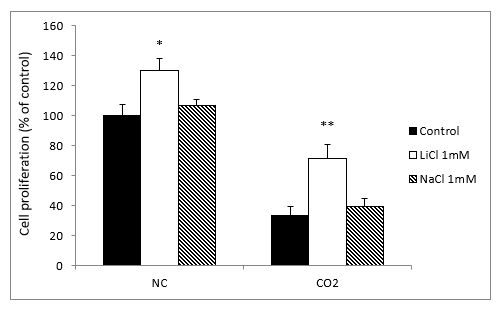
Figure 6: Effects of treatment with LiCl (1 mM) and NaCl (1 mM) after 72 hours exposure on SH-SY5Y cells proliferation in the absence, normoxia conditions (NC) or in the presence of CO2. The cell viability corresponds to the mean ± S.E.M of six independent experiments performed in triplicate. * P < 0.05 and ** P < 0.01 compared with control groups.
Western blotting
In relation to Western blots, the COX-2 antibody detected a prominent band at about 72 kDa. Under normoxic conditions, COX-2 protein levels, in the lithium-treated group, were not modified by the different concentrations. After CO2 exposure, we observed up-regulation of COX-2 in the control group and at lower concentrations of LiCl. However, this effect diminished as the LiCl concentration increased: at 1 and 5 mM it reached levels very similar to those obtained in the normoxic group (Figure 7A and 7B). No significant differences were observed in SH-SY5Y cells treated with rotenone or antimycin A (results not shown)
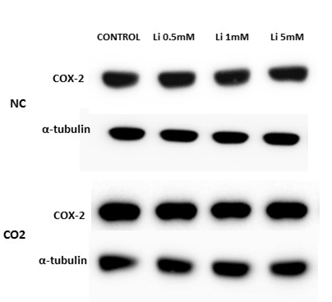
Figure 7A: Representative protein expression measured by immunoblotting of COX-2 in SH-SY5Y cells treated for 72 hours with different concentrations of LiCl (0.5-5 mM) in the absence, normoxia conditions (NC), or in the presence of CO2.
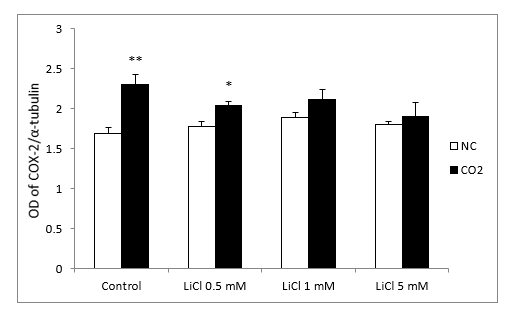
Figure 7B: Relative optical density (OD) of COX-2 to α-tubulin. All results are expressed as means ± S.E.M of six independent experiments performed in triplicate. *P < 0.05 and **P < 0.01 compared with normoxia conditions (NC) group.
Discussion
There are several reports that mood stabilizers such as lithium have neuroprotective properties and enhance resistance to oxidative stress against a wide array of insults, both in vitro and in vivo; but only a few studies have specifically examined the effects of lithium at therapeutic doses. Despite the conflicting reports, our results demonstrated that 72 hours of treatment with lithium was sufficient to observe a clear effect. Thus, at least in SH-SY5Y cell cultures, long-term treatment with lithium is not necessary to detect changes in parameters relating to oxidative stress.
HE fluorescence analysis was used to measure O2- production in different cells. HE reacts with O2- to form ethidium bromide, causing an increase in red fluorescence. This protocol has several advantages over other methods for detecting O2- produced in small amounts in different cells, as it allows for the continuous formation and accumulation of HE, which is stable in cells for prolonged periods of time [34]. Our results demonstrated a significant reduction in the production of O2- in the presence of all the lithium concentrations used, which shows the potential capacity of this ion to decrease O2- levels in SH-SY5Y cells. DCF was used to measure intracellular ROS production in various cells and there are authors who postulate that it could be suitable to measure H2O2 production [29]. Our results demonstrated that no significant differences were obtained in the intracellular ROS levels under standard conditions after LiCl treatment.
It is known that CO2 exposure induces redox status alterations, resulting from an increase in intracellular ROS generation. In particular, treatment of human neuroblastoma SH-SY5Y cells with CO2 causes an increase in intracellular ROS production that persists 24 h after CO2 exposure [26]. This could be related to the known property of CO2 to produce a hypoxic condition in cells and tissues. Levels of intracellular ROS increase under hypoxia in different cells [26, 35]. In addition, the reoxygenation of hypoxic cells can result in free- radical formation, sustained also by a hypoxia-induced decrease in cell antioxidant defenses [36]. To test the role of CO2-induced oxidative stress in our experimental model, we studied the production of O2- and intracellular ROS levels in the presence of CO2. Incubation with CO2 for 4 hours produced, after 24 hours of reoxygenation, a significant decrease in O2- and ROS production at all the concentrations of LiCl assayed. These results indicate that, in contrast to its effects on O2- levels, lithium only acts on intracellular ROS production, when the levels of this compound are altered by a factor than can damage the organism.
Several studies have suggested that the activity and expression of mitochondrial ETC components are altered in the brain of patients with bipolar disorder [30]. Complexes I and III are the main sites from which electrons are released and react with oxygen in intact cells, resulting in ROS production, thus causing oxidative stress [37]. Our results demonstrate that the inhibitors of mitochondrial complexes I and III, rotenone and antimycin A respectively, induced mitochondrial dysfunction as evidenced by the increased production of ROS and cell death. It has been suggested that mitochondria represent an important link between risk factors and eliminating injured mitochondria could be critical to prevent cells from enacting a series of pro-apoptotic cellular responses. In the presence of LiCl, the effects of rotenone decrease significantly, indicating that this ion protects the cells from ROS, albeit only partially. However, lithium is not capable of modifying the response to antimycin A; indicating that the response, in our experimental model, would be essentially related to mitochondrial complex I.
In relation to the results obtained with DCF, the inhibitor of complex I, rotenone, does not produced any difference from the control, in the presence of different concentrations of LiCl or its absence. This indicates that in contrast to what happened with O2-, at least in neuroblastoma cells, rotenone does not modify the production of intracellular ROS. It has been suggested that in HEK293 cells, ROS levels are not detectably increased by rotenone or antimycin A treatment. Our results suggest that very little intracellular ROS are formed or that it’s are effectively removed from mitochondria. This is in line with the hypothesis that mitochondria can act as cellular sinks of H2O2. [37, 38].
When the cells were treated with antimycin A, only the control and the lower concentration of LiCl significantly increase DCF production very slightly in comparison to the control group. Therefore, LiCl may not decrease the production of DCF induced by antimycin A in any case suggesting that, with respect to ROS production, lithium would seem to not affect the mitochondrial complexes I and III in SH-SY5Y cells. It is possible that other mitochondrial sites may contribute to ROS production [14]. In short, these results and those obtained under normoxia conditions suggest that exposure to low concentrations of lithium confer significant protection against oxidative stress. Consistent with our results, Frey et al. reported that lithium inhibited lipid peroxidation in the hippocampus of rats. Moreover, lithium treatment increases the antioxidant enzymes superoxide dismutase and catalase in several tissues, including several brain regions [16, 39].
Likewise, it has been demonstrated in vitro that lithium inhibits the increase of lipid peroxidation and protein oxidation induced by glutamate and decreases the vulnerability of neuronal cells to cell injury by increasing antioxidant defenses, perhaps from putative mitochondrial disturbances [6, 40, 41]. Not only was this protective effect found in animals and cell cultures, but in a recent study a decrease was found in the concentration of H2O2 in healthy humans following lithium treatment, associated with a decrease in oxidative stress [42]. In short, our results suggest that lithium treatment after CO2 exposure reduces the damage induced by hypoxia induced by CO2 in cell cultures; this protective effect could be due to a decrease of the oxidative products.
However, several studies suggest that lithium protects neurons from death induced by a wide array of neurotoxic insults and stimulates neurogenesis and the proliferation of glial and neuronal cells [7, 43, 44]. Likewise, other authors showed that in the presence of low concentrations of lithium, SH-SY5Y cells proliferated faster than controls and suggested that the phosphoinositol or Wnt pathway was involved in this stimulatory effect [6]. On the other hand, Nciri et al. also demonstrated that chronic exposure to low lithium concentration favored neurogenesis and brain regenerative capacity [28]. Our results support these hypotheses, especially after cell cultures were submitted to hypoxia by CO2 exposure. In this case, we observed a dose-dependent increase with LiCl in cell proliferation, whereas under normoxia conditions only a slight (but significant) increase was found.
In the presence of rotenone and antimycin A, the results were very similar to those obtained with CO2. Both rotenone and antimycin A significantly decrease cell proliferation while lithium increased it slightly but significantly. These findings indicate that at low concentrations, lithium could reverse cell apoptosis produced by different mechanisms and on its own, it is capable of stimulating cell proliferation, as observed in the control cells.
To verify the specificity of the effects of LiCl, we tested NaCl before and after the treatment with CO2. Unlike LiCl, NaCl does not modify intracellular ROS or O2- production and does not modify cell proliferation, indicating the specificity of lithium exposure in SH-SY5Y cell cultures. Our results also suggest that lithium does not modify COX-2 expression under normoxia conditions. After exposing the cultured cells to hypoxia with CO2, we observed a significant increase in the up-regulation of COX-2 in the control group, which dropped with increasing concentrations of lithium. In this regard, contradictory results have been reported. For example, several studies have demonstrated that lithium and other mood-modulating drugs significantly decrease expression of COX-2 in different experimental models, like primary cultured astrocytes, and in rat primary glia cells [40, 45]. Meanwhile, other studies have produced inconsistent results. For example, it was observed that the effect of lithium on COX-2 expression in different brain areas of rats increased COX-2 expression in the cerebral cortex and hippocampus but did not alter expression in other regions [46]. In contrast, Yuskaitis and Jope observed that lithium did not alter COX-2 expression in microglia cells [47]. In brief, those findings suggest that lithium reduces COX-2 expression in some tissues, but that it has no effect or enhances COX-2 expression in other tissues [48]. The use of different experimental models, different methods and different periods of chronic lithium treatment might explain these contradictory results. Our results showed that only under cell stress conditions does lithium slightly reduce COX-2 expression, but only to control values.
Conclusions
Lithium at very low concentrations partially protects SH-SY5Y cells against oxidative stress and corroborate the hypothesis that lithium, but not sodium, enhances the antioxidant defenses of various cells against cell death induced by oxidative stress. The protection afforded by lithium might be due, at least in part, to its inhibition of O2- from mitochondrial complex I; although it seems likely that multiple sites affected by lithium contribute to its antioxidant action. Also, lithium decreases the vulnerability of neuronal cells to cell injury. Meanwhile, exposure to CO2 produces up-regulation of COX-2 expression, while lithium reduces this at very low concentrations, suggesting that lithium does not act on COX-2 expression under normoxia conditions, but does under oxidative stress situations in neuroblastoma cells.
Conflict of Interest
The authors declare that they have not competing interests.
Article Info
Article Type
Research ArticlePublication history
Received: Fri 25, Jan 2019Accepted: Wed 13, Feb 2019
Published: Thu 28, Feb 2019
Copyright
© 2023 Frederic M?rmol Carrera . This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.NNB.2019.01.001
Author Info
Corresponding Author
Frederic M?rmol CarreraUnitat de Farmacologia, Facultat de Medicina, Universitat de Barcelona, Barcelona, Spain
Figures & Tables
References
- Marmol F (2008) Lithium: bipolar disorder and neurodegenerative diseases, possible mechanisms of the therapeutic effects of lithium. Prog Neuropsychopharmacol Biol Psychiatry 32: 1761-1771. [Crossref]
- Chen RW, Chuang DM (1999) Long term lithium treatment suppresses p53 and Bax expression but increases bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem 274: 6039-6042. [Crossref]
- Marmol F, Carbonell L, Cuffi ML, Forn J (1992) Demonstration of inhibition of cyclic AMP accumulation in brain by very low concentrations of lithium in the presence of α-adrenoceptor blockade. Eur J Pharmacol 226: 93-96. [Crossref]
- Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423: 435-439. [Crossref]
- Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, et al. (2002) Hsp27 as a negative regulator of cytochrome c release. Mol Cell Biol 22: 816-834. [Crossref]
- Allagui MS, Nciri R, Rouhaud MF, Murat JC, El Feki A, et al. (2009) Long-term exposure to low lithium concentrations stimulates proliferation, modifies stress protein expression pattern and enhances resistance to oxidative stress in SH-SY5Y cells. Neurochem Res 34: 453-462. [Crossref]
- Wada A, Yokoo H, Yanagita T, Kobayashi H (2005) Lithium potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J Pharmacol Sci 99: 307-321. [Crossref]
- Kim YH, Rane A, Lussier S, Ancerser JK (2011) Lithium protects against oxidative stress-mediated cell death in α-sinuclein-overexpressing in vitro and in vivo models of Parkinson’s disease. J Neurosci Res 89: 1666-1675. [Crossref]
- Rumia J, Marmol F, Sanchez J, Gimenez J, Carreño M, et al. (2013) Oxidative stress markers in the neocortex of drug-resistant epilepsy patients submitted to epilepsy surgery. Epilepsy Res 107: 75-81. [Crossref]
- Patel M, Li QY (2003) Age dependence of seizure-induced oxidative stress. Neuroscience 118: 431-437. [Crossref]
- Shin EJ, Jeong JH, Chuing YH, Kim WK, Ko KH, et al. (2011) Role of oxidative stress in epileptic seizures. Neurochem Int 59: 122-137. [Crossref]
- Morris G, Walder K, McGee SL, Dean OM, Tye SJ, et al. (2017) A model of the mitochondrial basis of bipolar disorder. Neurosci Biobehav Rev 74: 1-20. [Crossref]
- Toker L, Agam G (2014) Lithium, Inositol and Mitochondria. ACS Chem Neurosci 5: 411-412. [Crossref]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1-13. [Crossref]
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Bio 1: 304-312. [Crossref]
- Arraf Z, Amit T, Youdim MBH, Farah R (2012) Lithium and oxidative stress lessons from the MPTP model of Parkinson’s disease. Neurosci Lett 516: 57-61. [Crossref]
- Rao JS, Lee HJ, Rapoport SI, Bazinet RP (2008) Mode of action of mood stabilizers: is the arachidonic acid cascade a common target? Mol Psychiatry 13: 585-596. [Crossref]
- Rapoport SI (2001) In vivo fatty acid incorporation into brain phospholipids in relation to plasma availability, signal transduction and membrane remodeling. J Mol Neurosci 16: 243-261. [Crossref]
- Smith WL, Garavito DL, DeWitt DL (1996) Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Bio Chem 271: 33157-33160.
- Bosetti F, Rintala J, Seemann R, Rosenberg TA, Contreras MA, et al. (2002) Chronic lithium downregulates cyclooxygenase-2 activity and prostaglandin E2 concentration in rat brain. Mol Psychiatry 7: 845-850. [Crossref]
- Niwa K, Araki E, Morham SG, Ross ME, Iadecola C (2000) Cyclooxigenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci 20: 763-770. [Crossref]
- Yamamoto T, Nozaki-Taguchi N (1996) Analysis of the effects of cyclooxygenase (COX-1) and COX-2 in spinal nociceptive transmission using indomethacin, a non-selective COX inhibitor, and NS-398, a COX-2 selective inhibitor. Brain Res 739: 104-110. [Crossref]
- Chae SW, Kang BY, Hwang O, Choi HJ (2008) Cyclooxygenase-2 is involved in oxidative damage and alpha-synuclein accumulation in dopaminergic cells. Neurosci Lett 436: 205-209. [Crossref]
- Sharabi K, Lecuona E, Helenius IT, Beitel GJ, Sznajder JI, et al. (2009) Sensing, physiological effects and molecular response to elevated CO2 levels in eukaryotes. J Cell Mol Med 13: 4304-4318. [Crossref]
- Yu Y, Kuebler J, Groos S, Metzelder M, Kurpanik S, et al. (2010) Carbon dioxide modifies the morphology and function of mesothelial cells and facilitates transepithelial neuroblastoma cell migration. Pediatr Surg Int 26: 29-36. [Crossref]
- Montalto AS, Currò M, Russo T, Visalli G, Impellizzeri P, et al. (2013). In vitro CO2- induced ROS production impairs cell cycle in SH-SY5Y neuroblastoma cells. Pediatr Surg Int 29: 51-59. [Crossref]
- Pasquariello N, Catanzaro G, Marzano V, Amadio D, Barcaroli D, et al. (2009) Characterization of the endocannabinoid system in human neuronal cells and proteomic analysis of anandamide-induced apoptosis. J Biol Chem 284: 29413-29426. [Crossref]
- Nciri R, Desmoulin F, Allagui MS, Murat JC, El Feki A, et al. (2013) Neuroprotective effects of chronic exposure of SH-SY5Y to low lithium concentration involve glycolysis stimulation, extracellular pyruvate accumulation and resistance to oxidative stress. Int J Neuropsychopharmacol 16: 365-376. [Crossref]
- Jia Z, Misra HP (2007) Reactive oxygen species in in vitro pesticide-induced neuronal cell (SH-SY5Y) cytotoxicity: Role of NFκB and caspase-3. Free Radical Bio Med 42: 288-298.
- Andreazza, AC, Shao L, Wang JF, Young T (2010) Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry 67: 360-369. [Crossref]
- Hou L, Xiong N, Liu L, Huang J, Han C, et al. (2015) Lithium protects dopaminergic cell from rotenone toxicity via autophagy enhancement. BMC Neurosci 16: 82. [Crossref]
- Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Bio Chem 193: 265-275. [Crossref]
- Hansen MB, Nielsen SE, Berg K (1989) Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 119: 203-210. [Crossref]
- Peshavariya HM, Dusting GJ, Selemidis S (2007) Analysis of dihydroethidium fluorescence for the detection of intracellular and extracellular superoxide produced by NADPH oxidase. Free Radic Res 41: 699-712. [Crossref]
- Bentes de Souza AM, Wang CC, Chu CY, Briton-Jones CM, Haines CJ, et al. (2004) In vitro exposure to carbon dioxide induces oxidative stress in human peritoneal mesothelial cells. Hum Reprod 19: 1281-1286. [Crossref]
- Li C, Jackson RM (2002) Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol 282: 227-241. [Crossref]
- Forkink M, Basit F, Teixeira J, Swarts HG, Koopman W, et al. (2015) Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Bio 6: 607-616. [Crossref]
- Brown CG, Borutaire V (2012) There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 12: 1-4. [Crossref]
- Frey BN, Andreazza AC, Kunz M, Gomes FA, Quevedo J, et al. (2007) Increased oxidative stress and DNA damage in bipolar disorder: a twin-case report. Prog Neuropsychopharmacol Biol Psychiatry 31: 283-285. [Crossref]
- Shao L, Young LT, Wang JF (2005) Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry 58: 879-884. [Crossref]
- Wang HM, Zhang T, Li Q, Huang JK, Chen RF, et al. (2013) Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem Int 63: 345-353. [Crossref]
- Khairova R, Pawar R, Salvadore G, Juruena MF, de Sousa RT, et al. (2012) Effects of lithium on oxidative stress parameters in healthy subjects. Mol Med Rep 5: 680-682. [Crossref]
- Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM (2002) Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMD receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem 80: 589-597. [Crossref]
- Kim JS, Chang MY, Yu IT, Kim JH, Lee SH, et al. (2004) Lithium selectively increases neuronal differentiation of hippocampal neural progenitor cells both in vitro and in vivo. J Neurochem 89: 324-336. [Crossref]
- Nahman S, Belmaker RH, Azab AN (2012) Effects of lithium on lipopolysaccharide-induced inflammation in rat primary glia cells. Innate Immun 18: 447-458. [Crossref]
- Voutsinos-Porche B, Koning E, Kaplan H, Ferrandon A, Guenounou M, et al. (2004) Temporal patterns of the cerebral inflammatory response in the rat LiCl-pilocarpine model of temporal lobe epilepsy. Neurobiol Dis 17: 385-402. [Crossref]
- Yuskaitis CJ, Jope RS (2009) Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation induced neurotoxicity. Cell Signal 21: 264-273. [Crossref]
- Nassar A, Azab AN (2014) Effects of lithium on inflammation. ACS Chemical Neurosci 5: 451-458. [Crossref]