Immune Response Role of Angiogenesis Inhibitors
A B S T R A C T
Angiogenesis plays an important role in tumor growth. Established vasculature provides a supply of nutrients and other necessary survival factors for tumor cell maintenance. In addition, immune factors with capacity to both decrease immune activity leading to cancer suppression and to increase anticancer response are provided via VEGF stimulated angiogenesis. However, VEGF provides more than angiogenesis stimulation; it is itself a growth factor with activity to also decrease the stimulation of dendritic cells (DCs) and T cells involved in anti-cancer mechanisms. As such inhibition of VEGF provides immune therapeutic advantage. This was well demonstrated by IFN-ɣ ELISPOT assay in which T lymphocytes antitumor response was measured against multiple myeloma cells following exposure to myeloma lysate-loaded dendric cells. Block of VEGF lead to enhanced T lymphocyte anticancer immune response. Through stimulation of the immune system angiogenesis inhibitors can work in conjunction with immunotherapy, chemotherapy and/or radiation therapy. Recent clinical trials in advanced renal cell carcinoma, non-small cell lung cancer (NSCLC), and hepatocellular carcinoma have evidenced improved outcomes due to an immune enhancing effect with angiogenesis inhibition and in particular immune checkpoint blockade treatment.
Keywords
Angiogenesis, VEGF, autologous tumor vaccine, bevacizumab
Introduction
Angiogenesis is defined as the growth of blood vessels from a system of existing vasculature, and it is a process that occurs both physiologically and pathologically [1]. In physiology, angiogenesis is important for the function and formation of capillaries, which allow for the exchange of necessary metabolites and nutrients throughout the body. In this way, angiogenesis allows normal tissue as well as regenerating tissues access to a continuous supply of nutrients [2]. Being able to manipulate angiogenesis could have some therapeutic value, as stimulation of angiogenesis could be beneficial in ischaemic heart disease, peripheral arterial disease, and wound healing [1]. Any alterations in metabolic activity, as well as oxygen levels, can change the rate of angiogenesis.
Important differences exist between tumor vasculature and normal blood vessels. Tumor vessels in the tumor microenvironment (TME) are irregular shaped, disorganized, branched and leaky [3]. The basement membrane underlying endothelial cells is discontinuous [4]. Pericytes are specialized mesenchymal cells that coat and stabilize the endothelium of tiny blood vessels [5]. In the tumor microenvironment (TME), pericytes are loosely adhered to endothelium and their cytoplasmic projections invade parenchyma. Angiogenesis factors such as platelet derived growth factor (PDGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF) also play important roles which contribute to tumor growth and metastasis [6]. In a hypoxic tumor microenvironment (TME), VEGF is secreted in a paracrine fashion from pericytes which leads to increased endothelial cell proliferation [7]. VEGF reduces pericyte coverage on nascent vascular sprouts through inhibition of PDGFRβ signaling which leads to vessel destabilization [8]. Furthermore, pericytes participate in angiogenesis by increasing expression of membrane type 1 metalloproteinase (MT1-MMP) at the migrating tip of newly formed endothelial vessels leading to extracellular matrix degradation which facilitate the growth of new blood vessels, tumor invasion and metastasis.
Furthermore, growing tumors have a high need for nutrients related to constantly proliferating cells which are competing with each other for continued nutrient source, resulting in higher interstitial pressures and interfering with the diffusion of nutrients and metabolites to normal cells [2]. Angiogenesis factors in tumor cells give these cells the ability to survive and proliferate in this otherwise inhospitable environment [9]. In normal tissues, there is no pathological neovascularization as excessive angiogenesis is turned off, but due to the hostile environment in many tumors, angiogenesis becomes stimulated through tumor cell production of growth factors [10]. A variety of growth factors coordinate with VEGF to optimize angiogenesis and immune modulatory activity. Table 1 summarizes current interactive growth factors important in angiogenesis, angiogenesis related functions, and involvement in cancer with listed anticancer targeted treatment.
Table 1: Summary of important growth factors, function in angiogenesis, and current use in cancer therapy.
|
Growth Factors |
Known Function(s) |
Current Use(s) |
|
Agrin [11, 12] |
Induces the aggregation of nicotinic acetylcholine receptors; synaptic development, signaling in the brain, and plasticity [13] |
Sorafenib |
|
FAK-PyK2 inhibitor PF562271 |
||
|
Angiopoietins Astex FGFR Inhibitor
|
Angiogenesis; inflammation; maintains resting state of the endothelium [14] |
Neutralizing anti-Ang2 antibody in mice bearing xenografts of human A431 epidermoid tumor and Colo205 colon cancer [15] |
|
Ang2 + bFGF caused inhibition of angiogenesis in rat corneas [16] |
||
|
Fibroblast Growth Factor (FGF) |
Increases endothelial cell migration; promotes capillary morphogenesis |
TKI258 (dovitinib) Phase I trial in patients with advanced solid tumors [17] |
|
BMS-582664 (brivanib) targets VEGF-R2 and FGF-R1 and -2 [18] |
||
|
E7080 [19] |
||
|
BIBF 1120 (vargatef) targets VEGF receptors, FGF receptors, and PDGF receptors, especially in non-small cell lung cancer [20] |
||
|
AZD4547 inhibits FGFR tyrosine kinases 1, 2 and 3 [21] |
||
|
FP-1039 targeting FGFR2 in endometrial carcinoma [22] |
||
|
Platelet derived growth factor (PDGF) |
Activates autocrine and paracrine systems; encourages growth, survival, and motility in malignant, vascular, and stromal cells [23, 24]
|
Imatinib mesylate (PDGFR inhibitor) in a mouse model of cervical carcinogenesis slowed progression of premalignant lesions and impaired growth of invasive carcinoma [25] |
|
Transforming Growth Factor (TGF) |
Induce anchorage‐independent growth of target cells otherwise incapable of such growth [26] |
TGF-β inhibition in hepatocellular carcinoma, colorectal cancer, and glioblastoma multiforme xenograft [27-29] |
|
Galunisertib, (small molecule inhibitor of TGF-βRI) + orafenib, ramucirumab in hepatocellular carcinoma |
||
|
PF-03446962 (a monoclonal antibody against TGF-β) + regorafenib in colorectal carcinoma [30] |
||
|
Tumor Necrosis Factor-α (TNF-α) |
Inflammation; stimulate granulocyte-macrophage-colony stimulating factor (GM-CSF), interleukin-1 (IL-1), and angiogenic factors from other cells; induce endothelial cell differentiation [31] |
Golimumab inhibiting angiogenesis and growth in vivo in metastatic oral squamous cell carcinoma cells [32] |
|
Vascular endothelial growth factor (VEGF) |
Capillary morphogenesis; release of von plasminogen activator (PA), and plasminogen activator receptor (PA-R), Willebrand factor, integrins, and interstitial collagenase; increases vascular permeability and fenestration [33] |
Lung and colon cancer [34, 35] |
Another way in which growth factors such as bFGF, VEGF, EGF and PlGF play a role within tumors is their ability to prevent leukocyte migration and infiltration into tumor microenvironments through downregulation of adhesion molecules. These adhesion molecules include Intercellular Adhesion Molecule-1 (ICAM-1), Vascular Cell Adhesion Molecule (VCAM-1), and E-selectin which are necessary for leukocyte interaction in the vascular endothelium [10]. Therefore, the presence of angiogenic growth factors produced by the tumor results in suppression of these adhesion molecules and thus suppression of the adhesive properties within the endothelium. Thus, angiogenesis is required for tumor growth and metastasis formation [36].
It has also been found that endothelial cells which have been exposed to TNF and IFN-ɣ from tumors are unresponsive to tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), and interleukin-1 (IL-1) [10]. The combination of absent leukocyte adhesion receptors and the unresponsiveness to inflammatory mediators is called tumor endothelial cell ‘anergy’. Tumor endothelial cell anergy allows the tumor cell to escape immune surveillance in order to grow and metastasize [37]. Another way by which tumor growth is promoted involves leukocytes within a tumor. Leukocytes stimulate monocytes and macrophages, which in turn induce growth signals in tumor cells and therefore promote angiogenesis, which could lead to a worse prognosis [38, 39].
VEGF Signaling Mechanisms
In addition to placental growth factor (PGF), 32 proteins have been classified as angiogenesis factors, such as VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E [40, 41]. VEGF-A regulates angiogenesis. VEGF-C and VEGF-D play a major role in lymphangiogenesis [42]. VEGF-A undergoes alternative exon splicing resulting in multiple isoforms which include VEGF165, VEGF121, VEGF165, VEGF189, and VEGF206. Among these isoforms, VEGF165 (VEGF164 in mice) is the most important as it is the most prevalent and physiologically relevant isoform [43]. There are multiple receptors of VEGF (VEGFR) which include VEGFR-1 and VEGFR-2, predominantly expressed on endothelial cells. Ligands that bind to VEGFR-1 include VEGF-A, VEGF-B, and PGF. Ligands binding to VEGFR-2 includes VEGF-A, VEGF-C, and VEGF-D. Additionally, VEGFs also bind with high affinity to co-receptors such as neuropilin (NRP1 and NRP2) and to heparan sulfate proteoglycans (HSPGs). Furthermore, VEGF binds to auxiliary proteins, such as integrins and ephrin B2.
VEGFR receptor homodimerization or heterodimerization leads to activation of the tyrosine kinase and autophosphorylation of tyrosine residues in the receptor intracellular domains. VEGFR-1 is considered a negative regulator of VEGF. VEGFR-1 works as a decoy receptor that binds to PGF and prevents VEGF binding to VEGFR-2 [44]. Most of the physiological effects of VEGF-A are mediated through binding to VEGFR-2. VEGFR1–VEGFR-2 heterodimer mediates embryonic endothelium and atherosclerotic lesions. VEGFR-2 homodimer is present on the vascular endothelial cells. VEGF-A binds to the second and third extracellular immunoglobulin (Ig) loops of VEGFR-2, inducing receptor dimerization. VEGFR-2 downstream signaling pathway includes phospholipase Cγ (PLCγ), the ERK1/2 pathway, and the PI3K–AKT–mTOR pathway in addition to SRC and small GTPases. VEGF binding to VEGFR-2 leads to phosphorylation of specific tyrosine residue Y1173 in mice (corresponding to Y1175 in the human protein) in VEGFR-2, which results in the internalization of VEGFR-2 into early endosome antigen 1 (EEA1)-positive endosomes and Ca2+-dependent signaling in PLCγ and nuclear factor of activated T cells (NFAT) pathways. These signaling pathways lead to changes in gene expression responsible for cell migration and proliferation [45]. Briefly, PLCγ generates inositol 1,4,5‑trisphosphate (IP3) and diacylglycerol (DAG). IP3 releases Ca2+ from the endoplasmic reticulum. Ca2+ and DAG activate protein kinase Cβ2 (PKCβ2) which then regulates the RAF1–MEK–ERK1/2 cascade [46, 47].
Understanding VEGFR signaling mechanism is crucial to studying the effects of VEGF in the cell. As VEGFR is expressed in endothelial cells, increased VEGFR-2 signaling is responsible for angiogenesis including hypersprouting and hyperbranching of vasculature, as well as vascular permeability. As tumor cells are shown to express VEGFs, the signaling of this pathway can activate blood vessel growth around the source of the tumor to promote tumor proliferation. In addition, the growth of lymphatic vessels in lymphangiogenesis is modulated through VEGFR-3 activation. Tumor metastasis is mediated through lymphatic system through drainage of cells, so the upregulation of VEGFR-3 can lead to metastasis through the immune system [48].
Immune Effects of VEGF
Tumor cells evolve many mechanisms to evade recognition by the immune system. Tolerance toward tumor cell antigens could happen by decreasing stimulation of accessory molecules on lymphocytes, such as CD80 (B7.1) and CD86 (B7.2) [49]. Also, tumor cells may downregulate major histocompatibility complex (MHC) molecules to escape from cytotoxic T cell recognition [50]. Furthermore, tumors may produce immune inhibitory molecules such as IL-10, transforming growth factor-beta (TGF-β), or prostaglandins [10]. Other mechanisms involve activation of T-regulatory cells which have immunosuppressive effects [51, 52]. In addition, tumor cells can downregulate the expression of endothelial adhesion molecules like VCAM-1 and E-selectin that are necessary for interactions with leukocytes, such as granulocytes, macrophages, natural killer cells and lymphocytes [53].
However, for the immune response to affect malignant growth, the immune effector cells need access to the tumor microenvironment. Both VEGF and FGF influence microenvironment access [54, 55]. Pro-angiogenic cytokines modulate leukocyte adhesion molecules such as P-selectin, E-selectin, ICAM-1, and VCAM-1and may affect tumor anergy [10, 56]. With respect to leukocyte extravasation, P-selectin and E-selection are expressed on the surface of endothelium and enable a binding site for leukocytes circulating in the blood via interaction with the leukocyte adhesion protein Sialyl Lewis X [57, 58]. First entry into the extravascular space requires adherence to the endothelial surface and interaction of ICAM-1 and VCAM-1 with CD11/18 2-integrins [58, 59]. Upon interaction of Sialyl Lewis X with P- and E-selectins, CD11/18 2-integrins are activated on the leukocytes surface. Once this adhesion process has taken place, the leukocyte can transmigrate across the endothelial surface without damaging endothelial integrity. Expression of CD31, or Platelet/Endothelial Cell Adhesion Molecule 1 (PECAM-1) on the leukocytes surface facilitates this process (Figure 1) [60].
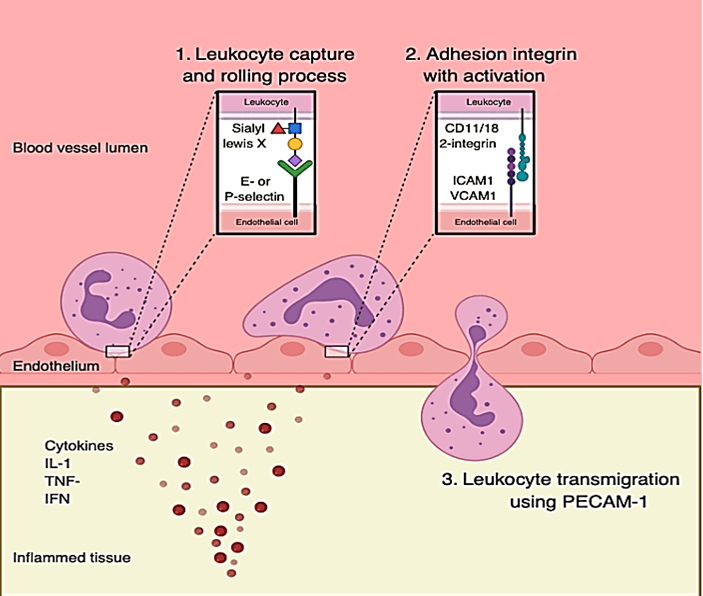
Figure 1: Leukocyte adhesion and migration process through endothelial cells.
However, in endothelial cells found in tumor areas, these extravasation molecules are largely absent, which makes it difficult for leukocytes to penetrate tumor cells through the vasculature [10]. Furthermore, pro-angiogenic factors inhibit TNF-α, IL-1 and IFN-ɣ which enhance expression of P-selectin and E-selectin on the endothelial surface [56].
IL-1, TNF-α, and IFN-γ also upregulate ICAM-1 and VCAM1 [57]. It has been shown that endothelial cells exposed to a growth-inducing mitogen such as bFGF first upregulate ICAM-1 followed by a marked and prolonged suppression of ICAM-1 expression [10, 61]. Many of the immune effects of VEGF are mediated by its action on dendritic cells (DCs). VEGF effects on DCs include the ability to inhibit the differentiation of DCs from hematopoietic stem cell CD34+ precursor, the functional maturation of immature-DCs, the antigen presenting function of mature DCs via MHC II and CD86 (B7-2), reduce DC ability to uptake antigens and differentiate into endothelial-like cells [62, 63]. These immune inhibitory functions of VEGF are reversed with VEGF inhibitors.
Gabrilovich et al. studied the effect of tumor cell supernatant on maturation of dendritic cells [62]. Human CD34+ cells from umbilical cord blood and were cultured with granulocyte-macrophage colony-stimulating factors (GM-CSF) and TNF-α to generate DCs. DCs were cultured in the presence of tumor supernatants derived from breast and colon adenocarcinomas cell lines and control fibroblasts. On day 12, cells were harvested, washed, and irradiated. They were then incubated with allogeneic control T cells and a significant reduction in the ability of the cells to stimulate T cells was observed. Also, CD34+ cells in the presence of tumor supernatant showed reduced ability to take up fluorescein isothiocyanate (FITC)-dextran which is a distinct feature of DCs. From this experiment, it was shown that VEGF production by tumor cells inhibits the functional maturation of dendritic cells [62].
Other studies have further characterized the observed effect on VEGF on DC maturation. The Yang et al. group explored the defective maturation and function of DCs in myeloma patients. They found that autologous tumor antigen loading to DCs negatively affected DC functional maturation, as indicated by the suppressed expression of maturation markers, altered cytokine secretion (IL-6, IL-10, IL-12), and decreased T cell stimulatory capacities and cytotoxic T cells generation. A study of bone marrow samples in addition to peripheral blood stem cell (PBSC) products from 6 multiple myeloma patients were collected after giving cyclophosphamide and G-CSF and achieving clinical response. Mononuclear cells were isolated, and DCs were generated from CD14+ monocytes and stimulated by GM-CSF and IL-4. DCs were then pulsed with myeloma cell lysates in the presence or absence of a neutralizing anti-VEGF-antibody. After the lysate pulsed DCs were cultured with an anti-VEGF neutralizing antibody, significantly decreased IL-6 and IL-10 secretion was observed. However, IL-12 secretion was significantly increased. DCs loaded with myeloma lysates at early or progressive states revealed decreased expression of CD80, CD83, CD86 and HLA-DR, in comparison with myeloma lysate-unloaded DCs. Through use of IFN-γ ELISPOT assay, cytotoxic T lymphocytes (CTLs) response was measured. DCs were generated with or without loading of myeloma lysates in the presence or absence of anti-VEGF antibody. It was observed that myeloma lysate-pulsed DCs treated with anti-VEGF antibody resulted in a higher number of IFN-γ-secreting CTLs compared to DCs loaded with myeloma lysates without anti-VEGF antibody. It was concluded that the addition of anti-VEGF enhanced the DC maturation and increased DC ability to stimulate cytotoxic T cell response [64].
The underlying mechanism of VEGF-A effect on dendritic cells is through inhibition of nuclear factor kappa-light chain-enhancer of B cell (NF-κB) signaling. VEGF-A inhibits maturation of DCs into functional cells by phosphorylating transcription factors such as STAT3 and ERK. These phosphorylated products inhibit NF-κB signaling [64]. For example, phosphorylated ERK results in activation, which can increase the production of IL-10 and suppress IL-12, and this ultimately results in the suppression of NF-κB signaling and decreases the maturation of DCs [65, 66]. C-Rel signaling in the NF-κB pathway is shown to be linked with decreased immune signaling and DC maturation with decreased IL-12, and VEGF inhibition has been shown to suppress c-Rel signaling [67].
The effect of angiogenesis inhibitors on the immune system can be further illustrated through a VEGFR inhibitor such as axitinib which has been FDA approved for second line therapy of metastatic renal cell carcinoma. It acts selectively on VEGF and has been shown to have anti-angiogenic and immune-modulatory functions [67, 68]. Axitinib also interferes with lipopolysaccharide’s (LPS) effect on the upregulation of DC activation markers CD80, CD83, and CD86, which results in a decrease in the expression of these activation markers through inhibition of the toll-like receptor (TLR) 4 pathway. Axitinib also resulted in significant reduction in interleukin-12 level (IL-12 is an important cytokine secreted by DCs and results in T cell activation) and TNF-α. The mechanism underlying the effect of VEGF on DC maturation was via phosphorylation of p38 and STAT3. The addition of axitinib resulted in a dose dependent decrease in phosphorylation of p38 and STAT3.
Pre-clinical studies using axitinib in melanoma also showed promising results. For example, Bose et al. studied the combination of axitinib with peptide-based vaccination using a subcutaneous B16.OVA melanoma mouse model. Combination therapy resulted in extended survival with 40% of mice surviving 80 days post-tumor inoculation, whereas mice receiving monotherapy of vaccine or axitinib did not survive longer than 50 days post-tumor inoculation. Kaplan-Meier curve showed a higher survival of the combination therapy of vaccine + axitinib when compared to monotherapy (P <0.05) [69]. The inhibition of VEGF activity by anti-VEGF neutralizing antibody or anti-VEGF receptor antibodies overcome the functional inhibition of DCs [70]. The effect of VEGF-A is mediated by its binding to the VEGFR-1 receptor. Interestingly, the same receptor is expressed in CD34+ hematopoietic progenitor cells (HPCs). VEGF acts as a chemoattractant to HPCs. HPCs are hematopoietic progenitor precursor cells with the ability to differentiate into mature immune cells like neutrophils, dendritic cells, and macrophages. In addition, VEGF-A contributes to increase tumor vascularity by inducing endothelial cell differentiation from HPCs. Furthermore, VEGF-A inhibits NF-κB, thus preventing HPCs differentiation into mature immune cells [71].
DCs derived from peripheral monocytes can also differentiate into endothelial-like cells (ELCs) in the presence of VEGF. The differentiation into ELCs is detected by expression of ELC markers which include vWF, CD144 (VE-cadherin), CD105 (endoglin), acetylated low-density lipoprotein (AC-LDL)-receptor, CD36 (thrombospondin receptor), FLT-1 (VEGF receptor-1), and weaker expression of KDR (VEGF receptor-2). In addition, DC markers such as CD1a and CD83 are downregulated [72]. ELCs were able to form tube like structures when cultured in endothelial cell growth medium in the presence of VEGF, bFGF and TNF-α. The close proximity of DCs to blood vessels and their ability to differentiate into endothelial cells in the presence of tumor secreted VEGF by forming tube like structures play an important role in the neovascularization in the tumor microenvironment [73].
Along with DCs, VEGF also has the ability to regulate T cell activation. With increased signaling of VEGF-A and subsequent VEGFR interaction, decreased T cell function is noted. VEGF-A has been observed to enhance programmed cell death (PD-1) expression. PD-1 is an inhibitory checkpoint which leads to the dysfunction of T cells involved in antitumor immune responses. With specific blocking of VEGF-A and VEGFR, CD8+ T cells have been noted to result in decreased levels of PD-1. Additionally, tumors lacking VEGF expression have decreased PD-1 expression as well. The VEGF blockade can lead to upregulation of anti-tumor T cell response without inhibition of the pathway, as blockade of VEGF also results in increased IFN-γ levels [74]. VEGFR-1, specifically, has been associated with T cell signaling as well. With increased release of VEGF from tumor cells, VEGFR-1 signaling increases leading to production of IL-10 which can also decrease the response of anti-tumor T cells, similar to PD-1 signaling [75]. The tumor mechanisms have therefore been well developed to evade the immune system through inhibition of T cell activity. As VEGF decreases anti-tumor immune activity through DCs and T cells, the signaling of VEGF can be inhibited to improve immune responses in cancer [76].
Angiogenesis Inhibition for Cancer Therapy
There are different classes of Food and Drug Administration (FDA) approved angiogenesis inhibitors for cancer therapy that utilize different mechanisms. One of the classes is made up of endogenous inhibitors which are released from the extracellular matrix in order to prevent a response in the endothelial cells from angiogenesis inducers, such as VEGF, bFGF, IL-8, and PGF [77, 78]. Endogenous anti-angiogenesis inhibitors include endostatin, angiostatin and interferon-α (IFN-α) [79]. Endostatin, carboxy-terminal fragment of collagen XVII, is one of the promising anti-angiogenesis inhibitors. It works by inducing apoptosis in endothelial cells, inhibition of endothelial cells migration, and interfering with endothelial cell adhesions. Angiostatin, induces apoptosis in endothelial cells in addition to also act by inhibiting VEGF and basic fibroblast growth factor (bFGF) and also decreases endothelial cell migration [79]. IFN-α has been used to treat hemangiomas, refractory giant cell tumors, and angioblastomas. The main target of direct endogenous inhibitors of angiogenesis is VEGF which induces proliferation of vascular endothelial cells needed for blood vessels to induce tumorigenesis [77, 78].
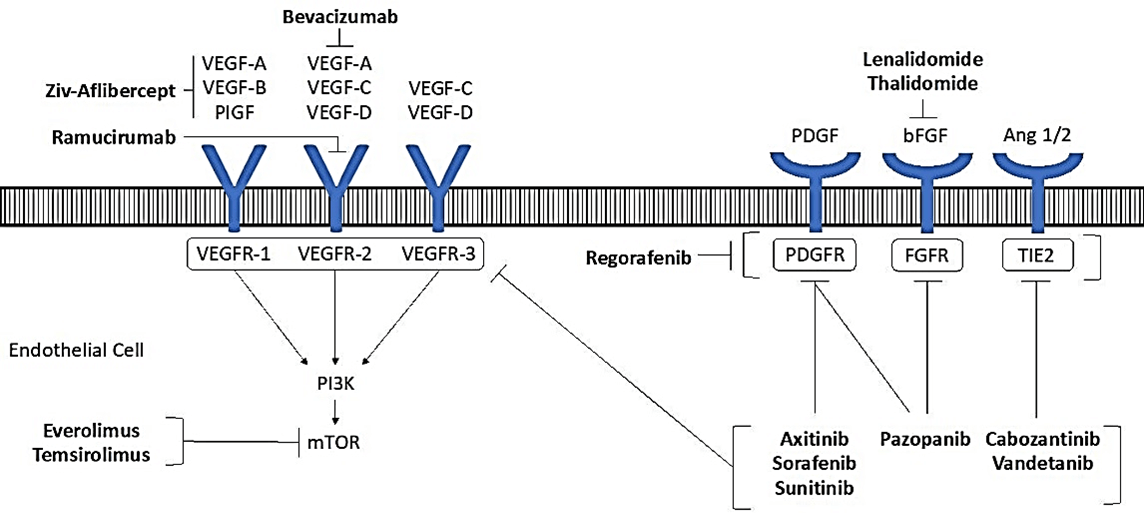
Figure 2: Signaling mechanism of angiogenesis inhibitors including the inhibition of VEGF and interaction with receptor to activate the signaling cascade.
Table 2: FDA approved angiogenesis inhibitors as cancer therapeutics (adapted from multiple sources including Yang 2017, Rajabi and Mousa 2017, and Ye 2016) [80-82].
|
Mechanism of Action |
Specific targets |
Generic Name (Brand Name), Year of FDA approval |
Indication(s) |
|
Anti-VEGF antibody |
VEGF-A |
Bevacizumab (Avastin®), 2004 [83] |
Colorectal, non-small-cell lung cancer (NSCLC), glioblastoma multiforme, and epithelial ovarian cancer |
|
VEGF inhibitor (trap mechanism) |
VEGF-A, VEGF-B, PlGF |
Ziv-aflibercept (Zaltrap®), 2012 [84] |
Colorectal cancer |
|
VEGFR antibody |
VEGFR-2 |
Ramucirumab (Cyramza®), 2014 [85, 86] |
Stomach cancer, gastroesophageal junction adenocarcinoma, NSCLC, and hepatocellular carcinoma |
|
Tyrosine kinase inhibitor (TKI) |
VEGFRs, PDGFRs, cKIT |
Axitinib (Inlyta®), 2012 [87] |
Renal cell carcinoma |
|
TKI |
VEGFRs cKIT, DDR2, FGFRs, FLT3, FMS, MUSK, cRAF, PDGFRs, Ret, TAO2 |
Sorafenib (Nexavar®), 2005 [88] |
Renal cell carcinoma and hepatocellular carcinoma |
|
TKI |
VEGFRs ARK5, CaMKIIs, CHK2, cKIT, cRAF, FGFR1, Flt3, FMS, Mer, PDGFR, Ret, TrkA |
Sunitinib (Sutent®), 2006 [89] |
Renal cell carcinoma, gastrointestinal carcinoma and PNETs |
|
TKI |
VEGFRs, cKIT, FMS, FGFR2, FLT3, MLK1, PDGFR |
Pazopanib (Votrient®), 2009 [90, 91] |
Renal cell carcinoma and advanced soft tissue sarcoma |
|
TKI |
VEGFRs PDGFRs, FGFRs, Tie2, DDR2, Trk2A, Eph2A, RAF-1, STK5 |
Regorafenib (Stivarga®), 2012 [92] |
Colorectal cancer, gastrointestinal stromal tumor and hepatocellular carcinoma |
|
TKI |
VEGFRs, cMET, Ret, cKIT, Axl |
Cabozantinib (Cometriq®), 2012 [86, 93] |
Thyroid cancer, renal cell and hepatocellular carcinoma |
|
TKI |
VEGFRs EGFRs, Ret |
Vandetanib (Caprelsa®), 2011 [94] |
Thyroid cancer |
|
bFGF inhibitor |
FGF |
Lenalidomide (Revlimid®) [95, 96] |
Myeloma, mantle cell, follicular and marginal zone lymphoma |
|
bFGF inhibitor |
FGF |
Thalidomide (Synovir, Thalomid®) [95-98] |
Myeloma
|
|
mTOR inhibitor |
mTOR |
Everolimus (Afinitor®) [99] |
Renal cell carcinoma, advanced breast cancer, pancreatic neuroendocrine tumors (PNETs), renal angiomyolipoma, and subependymal giant cell astrocytoma |
|
mTOR inhibitor |
mTOR |
Temsirolimus (Torisel®) [99] |
Renal cell carcinoma |
The second class of angiogenesis inhibitors is comprised of indirect inhibitors of angiogenesis which block the expression or activity of angiogenesis inducers such as VEGF. After the discovery of VEGF in 1989, several clinical trials involving angiogenesis inhibitors in cancer treatment resulted in a series of drugs to be approved by the FDA for use in cancer patients with bevacizumab among the first in 2004 [80]. The mechanisms of these drugs are described in (Figure 2) to show the interactions with the receptors [6]. A list of FDA approved angiogenesis inhibitors categorized by mechanism is provided with indication for cancer treatment in (Table 2) [81, 82]. Given the role of angiogenesis inhibitor and immune function enhancement attributed to use of angiogenesis inhibitors and it is unclear what proportion of angiogenesis inhibitors vs immune inhibitor provides anticancer effect in individual patients.
One of the most studied angiogenesis inhibitors in cancer therapy is bevacizumab (Genentech/Roche, San Francisco, CA) [40, 77, 100]. Bevacizumab is a humanized monoclonal antibody against all isoforms of VEGF-A. It was the first VEGF inhibitor approved for the treatment of cancer and is approved for the treatment of colorectal, non-small-cell lung, breast, renal cell cancers, and glioblastoma [101]. More recently, bevacizumab has also been approved for treatment in ovarian cancer [83]. Bevacizumab was initially approved for the treatment of metastatic colorectal cancer (mCRC). Hurwitz et al. demonstrated that bevacizumab in combination with irinotecan, bolus fluorouracil, and leucovorin the response rate (44.8% vs. 34.8% HR 0.62; P=0.004), progression-free survival (10.6 vs. 6.2 months; HR 0.54; P<0.001) and overall survival (20.3 vs. 15.6 months; HR 0.66; P<0.001) were significantly higher compared to patients given leucovorin/placebo [102].
The role of bevacizumab as adjuvant therapy with FOLFOX4 derived from Folinic acid (leucovorin) "FOL", Fluorouracil (5-FU) ‘F’, and Oxaliplatin (Eloxatin) ‘OX’ was further studied in previously treated mCRC patients with the comparison groups receiving monotherapy of FOLFOX4 or bevacizumab. Patients (n=829) were divided into three cohorts and results showed significantly improved survival with the addition of bevacizumab to FOLFOX4, including RR (22.7% for FOLFOX4 in combination with bevacizumab vs. 8.6% FOLFOX4 vs. 3.3% bevacizumab; P<0.0001, PFS (7.3 for FOLFOX4 in combination with bevacizumab vs. 4.7 FOLFOX4 vs. 2.7 months bevacizumab; HR 0.61; P<0.0001) and OS (12.9 for FOLFOX4 in combination with bevacizumab vs. 10.8 FOLFOX4 vs. 10.2 months bevacizumab; HR=0.75; P=0.0011) [103].
Saltz et al. showed significantly improved progression-free survival (PFS) of 9.4 months with bevacizumab in combination with FOLFOX or XELOX (oxaliplatin and capecitabine) as a first-line regimen in patient with colorectal cancer vs 8 months (HR 0.83; 97.5% CI, 0.72 to 0.95; P=0.0023) with placebo plus chemotherapy FOLFOX or XELOX [104]. There are also indirect angiogenesis inhibitors that have been FDA approved, including tyrosine kinase inhibitors (TKIs) that target the endothelial growth factor receptor (EGFR). Sorafenib and sunitinib are TKIs which block VEGF from interacting with EGFRs to prevent angiogenesis [77, 100]. Sunitinib (Sutent; Pfizer, New York, NY) is a multikinase inhibitor that inhibits multiple receptors including VEGFR-1, 2, 3, PDGFR, c-Kit, and RET. Sorafenib (Nexavar; Onyx and Bayer, San Francisco, CA) is a multikinase inhibitor that also inhibits multiple receptors including VEGFR-1, 2, 3, PDGFR, c-Kit, RET, and Raf [101]. Another TKI is pazopanib (Votrient; GlaxoSmithKline, Brentford, England) that inhibits VEGFR-1, 2, 3, PDGFR, and c-Kit. Sunitinib, sorafenib, and pazopanib have been approved as monotherapies in treatment of renal cell carcinoma and gastrointestinal carcinoma [101].
Temsirolimus (Torisel; Pfizer, New York, NY) and everolimus (Afinitor; Novartis, Basel, Switzerland) are rapamycin (mTOR) inhibitors which are considered to be another class of angiogenesis inhibitors involved in the PI3K-AKT-mTOR pathway [40]. This pathway is responsible for metabolism, growth, and proliferation found in tumor cells through activation of VEGF-associated angiogenesis and cell survival (survivin) [81, 99]. Angiogenesis inhibitors also include lenalidomide and thalidomide (Revlimid and Synovir; Celgene, Summit, NJ) which inhibit secretion of basic fibroblast growth factor (bFGF) and block E3 ubiquitin ligase from degradation of cereblon, and the complex of cereblon and E3 ubiquitin ligase then marks proliferation transcription factors for degradation [95-98, 105, 106].
Although angiogenesis inhibitors have been developed to treat various types of cancers, adverse effects are still present with treatment. Some of the side effects include hypertension, arterial thromboembolism, cardiac ischaemia, and cardiac dysfunction [107-109]. Renal side effects also include proteinuria and glomerulonephritis [108, 109]. Bevacizumab, in particular, has been associated with Grade 3 or 4 adverse events of hypertension, proteinuria, bleeding and impaired wound healing [110, 111]. With application of angiogenesis inhibitors in clinical treatment, adverse effects should be monitored closely. Though safety considerations are present, angiogenesis inhibitors can continue to be cautiously used in the treatment of cancer.
Synergistic Effects of Angiogenesis Inhibitors
Patients treated with angiogenesis inhibitors have shown greater T cell infiltration, number of leukocytes, and overall anti-tumor immunogenic response [10, 56]. Furthermore, because of its immunomodulatory response, anti-angiogenic drugs, such as bevacizumab, and immunotherapy drugs, such as PD-L1 inhibitors, have shown positive in vitro and in vivo responses in several different types of tumors [112]. Anti-angiogenic drugs have also been shown to have synergistic effects with chemotherapy medications, both in ability to reduce interstitial pressure within the tumor and as a drug delivery system for chemotherapeutic agents [55, 113, 114]. This facilitates broad use of angiogenesis inhibitors in combination both with immune therapeutics and more traditional chemotherapy.
It has also been shown that anti-angiogenic agents can work synergistically with radiation treatment [115]. One of the mechanistic features of radiation therapy is the ability to induce free radicals via reactive oxidative species (ROS) which leads to cellular apoptosis. Therefore, sufficient oxygenation must be present in cancer cells in order for radiotherapy to be effective [116]. Anti-angiogenic drugs such as bevacizumab can assist with this increased oxygenation process [111, 115]. It has been theorized that anti-angiogenic drugs block “immature” vessel growth and normalize angiogenesis towards a tumor. This, in turn, actually increases oxygenation in the tumor during the first four days of treatment. Therefore, when radiation therapy is combined with anti-angiogenesis during the oxygenation window, which is to say that bevacizumab is administered before radiation occurs, tumor growth has been shown to be delayed [115]. In sum, while anti-angiogenic therapy is effective on reducing tumor growth on its own, it can act synergistically when combined with immunotherapeutic, chemotherapeutic, or radiotherapeutic agents.
Radiation therapy may also increase neoantigen visibility through cell death, or when used at sub-lethal doses may result in genotypic changes which result in more neoantigens [117]. Vaccination also increases neoantigen display and may represent an attractive combination with angiogenesis inhibitors. For instance, Vigil, an autologous tumor vaccine that expresses GM-CSF and knocks down expression of furin has shown promising clinical benefit [118-124]. Vigil also increases the number of CD3+CD8+ T cells in peripheral blood [125]. Phase II results in advanced ovarian cancer patients revealed improvement in both relapse free and overall survival in the BRCA wild type population [126]. This effect may be attributed to clonal neoantigen display via MHC II expression which is maximized in the BRCA wild type population due to intact homologous recombination compared to the BRCA mutant population [127]. MHC II expression is key to Vigil activity, thus VEGF inhibition which increases MHC II expression would be logical. Combination of Vigil to increase neoantigen display and circulating T cells and angiogenesis inhibitors which increase MHC II expression, dendritic cell activation and maturation as well decrease interstitial pressure to allow T cell infiltration into the tumor microenvironment may facilitate an enhanced immune response.
Clinical Trials Supporting Immune Enhancing Effects of Angiogenesis Inhibitors
I Advanced Renal Cell Carcinoma
As immune enhancing effects were noted with angiogenesis inhibitors, combination with immunotherapy could provide optimal results for cancer patients. Immune checkpoint blockade therapies include PD-1 antibodies that increase anti-tumor T cell activity without tumor evasion of the immune response [128, 129]. For advanced renal cell carcinoma (aRCC), approved therapies include monotherapy of nivolumab, a PD-1 inhibitor, and sunitinib, a VEGF TKI. Sunitinib has shown immune modulatory effects in enhancing T cell activity in aRCC. Therefore, clinical studies have been developed to explore enhanced anti-tumor immune activity with angiogenesis and PD-1 inhibitors [128]. A Phase I study for aRCC was conducted to test the efficacy of nivolumab in combination with sunitinib or pazopanib. Safety assessment yielded adverse events in 100% of patients with Grade 3/4 treatment related AEs in 82% of patients [128]. Adverse events and toxicity have limited development of this combination and indicate that careful dosing and which immune inhibitor selected may be important.
Therefore, pembrolizumab in combination with axitinib was investigated in the KEYNOTE‑426 Phase III trial versus monotherapy sunitinib. At 12 months, OS was 89.9% and 78.3% (HR 0.54 95% CI: 0.38-0.74 P<0.0001) with combination therapy and standard treatment respectively. The PFS was 15.1 months and 11.1 months (HR 0.69; CI 0.57-0.84 P<0.001) and the ORR was 59.3% and 35.7% (95% CI 31.1 to 40.4 P<0.001) after a median follow-up of 12.8 months which again indicates combination pembrolizumab and axitinib is efficacious. Grade 3 or 4 adverse events were reported at a rate of 75.8% in combination therapy which can be comparable to 70.6% in monotherapy [130]. An important biomarker in predicting clinical response of axitinib and pembrolizumab treatment includes PD-1 (PD-1) expression suggesting enhanced anti-tumor immune activity, but the treatment showed significant improvement regardless of PD-L1 expression in patients [129, 130]. As a result of the Phase III trials, the combination therapy of pembrolizumab and axitinib was approved by the FDA in 2019 as first-line treatment for aRCC. This approval highlights the potential of combining angiogenesis inhibitor therapy with immune checkpoint blockade to improve patient outcomes.
Continued support of angiogenesis inhibition with immunotherapy has been shown in another combination approval by the FDA in 2019. Avelumab, an anti-PD-L1 monoclonal antibody, plus axitinib was compared with standard monotherapy of sunitinib in a Phase III trial with 886 aRCC patients. The median PFS for combination therapy was 13.8 months compared to monotherapy of 8.4 months (HR: 0.69; 0.56-0.84, 95% CI; P<0.001). The ORR was 51.4% (95% CI 46.6-56.1) for combination therapy and 25.7% (95% CI 21.7-30.0) for sunitinib only. In patients with PD-L1 positive tumors, the progression-free survival was similar to the overall population (HR 0.61 95% CI 0.47-0.79 p<0.001). In regard to adverse events, avelumab and axitinib led to 3 patient (0.7%) deaths due to toxicity whereas sunitinib treatment resulted in 1 patient death (0.2%). Similar rates of adverse events were recorded in the avelumab/axitinib group compared to sunitinib monotherapy. The exhibited efficacy and similar rate of toxicity to standard of care sunitinib indicates combination avelumab and axitinib can be beneficial for patients with aRCC [131]. Furthermore, increased evidence of synergistic effects of angiogenesis inhibition and immune checkpoint blockade emphasizes the importance continued research.
Further promising results were exhibited in the IMmotion150 study, a Phase II study in aRCC, combining bevacizumab with atezolizumab, an anti-PD-L1 inhibitor. After a median survival follow-up of 20.7 months, results indicated a median progression-free survival (PFS) of 11.7 (HR 1.00 95% CI 8.4-17.3; p 0.982) months with combination of bevacizumab and atezolizumab, while monotherapy of atezolizumab was only 6.1 (HR 1.19 95% CI 5.4-13.6 p 0.358) months. While results were not significant in the overall population, greater benefit was seen in PD-L1+ patients. The combination therapy median PFS was 14.7 (HR 0.64 95% CI 8.2-25.1 p 0.095) months compared to atezolizumab monotherapy of 5.5 (HR 1.03 95% CI 3.0-13.9 p 0.917) months. PD-L1 expression indicates CD8 T-effector cell and interferon gamma activity within the tumor. The addition of a VEGF angiogenesis inhibitor was observed to increase the anti-tumor immune response associated with decreased myeloid-derived suppressor cells responsible for evading the immune system [132]. Tissue analysis of aRCC patients treated with bevacizumab and atezolizumab showed increased T-helper 1 (TH1) chemokines along with CD-8 T-effector cells and natural killer cells contributing to the robust immune response [133]. Further analysis of angiogenesis and immune checkpoint inhibitors could definitively identify immunostimulatory effects of angiogenesis inhibitors for tumors building resistance to immunotherapy in aRCC as well as other types of cancers [132].
II Non-Small Cell Lung Cancer
Similar to studies seen in aRCC, advances in metastatic non-squamous non-small cell lung cancer (NSCLC) have also been made using anti-angiogenic and immunomodulatory combination therapy. In a Phase III study, 336 patients who were treated with atezolizumab, bevacizumab, carboplatin and paclitaxel were compared with 336 patients treated with bevacizumab, carboplatin and paclitaxel. The PFS was significantly improved with atezolizumab (HR 0.62; 8.3 vs 6.8 months; P<0.001). Subgroup analysis of EGFR mutations or anaplastic lymphoma kinase (ALK) translocations, KRAS mutations, low or negative PD-L1 expression, and liver metastases exhibited increased PFS with the addition of atezolizumab to bevacizumab and chemotherapy [134]. As previous Phase II and III trials with atezolizumab and chemotherapy combination did not show significant improvement in PFS compared to standard treatment of chemotherapy, the improved survival can be associated with the combination atezolizumab and bevacizumab from immunostimulatory effects [134-136]. Furthermore, analysis of patients in subgroups EGFR+ mutation and baseline liver metastases of the same Phase III clinical trial with continued enrollment resulted in no overall survival benefit between the atezolizumab and bevacizumab groups when treated with chemotherapy. However, improved outcomes were noted with the combination of atezolizumab and bevacizumab to chemotherapy which illustrated the enhanced interactions of angiogenesis inhibitors with immune checkpoint inhibitors, especially when EGFR was involved in tumorigenesis [137].
III Hepatocellular Carcinoma
The combination of atezolizumab and bevacizumab is also noteworthy in unresectable hepatocellular carcinoma. In a Phase Ib multicenter study, 60 patients were assigned to atezolizumab plus bevacizumab and 59 to atezolizumab monotherapy. Median PFS was 5.6 months for the combination therapy while monotherapy was 3.4 months (HR 0.55; 80% CI 0.40, 0.74 p=0.011), indicating the addition of bevacizumab improved survival outcomes. However, the combination therapy also posed serious Grade 3-4 adverse events such as hypertension and proteinuria in 7 (12%) patients, compared to 2 (3%) patients in the monotherapy cohort [138]. Similar results were observed in a Phase III study of atezolizumab plus bevacizumab versus sorafenib in unresectable hepatocellular carcinoma. PFS was improved in the combination therapy group (6.8 months) versus the sorafenib group (4.3 months) (HR 0.59; 95% CI: 0.47-0.76; P<0.001). The most common Grade 3 or 4 adverse event was hypertension which was present in 15.2% of the combination therapy group [139].
Future Directions
The clinical trials discussed in the previous section show the wide array of combination therapy, including anti-angiogenesis and immune checkpoint blockade, that have been studied in aRCC, NSCLC and hepatocellular carcinoma. Similar combination therapy development is recommended to continue for other types of cancers as well. Furthermore, the specific combinations that have been FDA approved include avelumab, an anti-PD-L1 monoclonal antibody, and pembrolizumab, an anti-PD-1 monoclonal antibody, with axitinib, VEGFR inhibitor, for aRCC. Therefore, continued development of combination therapy should focus on therapeutics with similar mechanisms to anti-PD-L1 and anti-PD-1 inhibition along with VEGF inhibition.
Recycling of previous combination therapies is also important to consider. Since chemotherapy has previously shown efficacy with angiogenesis, the use of chemotherapy can be emphasized with both immunotherapy and anti-angiogenic therapy. As evidenced in the Phase III trial for NSCLC patients, the synergistic effects in addition to chemotherapy have shown significant overall improvement in patients receiving atezolizumab plus bevacizumab plus carboplatin plus paclitaxel (ABCP). Improved median overall survival was reported in the intention-to-treat population for the combination of ABCP at 19.8 months (95% CI: 17.4-24.2) whereas BCP median overall survival was 14.9 months (95% CI: 13.4-17·1) (HR 0.76; 95% CI: 0.63-0.93) [137]. With the significant improvement evidenced, chemotherapy should continue to be considered in development of research trials including anti-angiogenesis inhibition and immune checkpoint blockade to optimize results for patients.
Conclusion
Through the years, angiogenesis inhibition has been studied meticulously, leading to the evolution of anti-angiogenesis cancer therapeutics with FDA approval. In this paper, we discussed angiogenesis in the tumor microenvironment and signaling pathways involved with emphasis on FDA approved angiogenesis inhibitors. We have showed that angiogenesis inhibitors enhance immune response toward tumor cells. These mechanisms could be harnessed alongside newer immunomodulatory drugs to improve cancer treatment. Further clinical trials should be developed to assess benefits and risks. In addition, biomarkers for an enhanced response can also be significant in identifying patients and cancer types suitable for combined anti-angiogenesis and immunomodulatory treatment. The promising results of this coupled therapy also highlights the potential for other combinations including chemotherapy that need to be investigated in the field of cancer therapy.
Article Info
Article Type
Research ArticlePublication history
Received: Thu 07, Jan 2021Accepted: Sat 23, Jan 2021
Published: Mon 01, Feb 2021
Copyright
© 2023 John Nemunaitis. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.COR.2021.02.01
Figures & Tables
Table 1: Summary of important growth factors, function in angiogenesis, and current use in cancer therapy.
|
Growth Factors |
Known Function(s) |
Current Use(s) |
|
Agrin [11, 12] |
Induces the aggregation of nicotinic acetylcholine receptors; synaptic development, signaling in the brain, and plasticity [13] |
Sorafenib |
|
FAK-PyK2 inhibitor PF562271 |
||
|
Angiopoietins Astex FGFR Inhibitor
|
Angiogenesis; inflammation; maintains resting state of the endothelium [14] |
Neutralizing anti-Ang2 antibody in mice bearing xenografts of human A431 epidermoid tumor and Colo205 colon cancer [15] |
|
Ang2 + bFGF caused inhibition of angiogenesis in rat corneas [16] |
||
|
Fibroblast Growth Factor (FGF) |
Increases endothelial cell migration; promotes capillary morphogenesis |
TKI258 (dovitinib) Phase I trial in patients with advanced solid tumors [17] |
|
BMS-582664 (brivanib) targets VEGF-R2 and FGF-R1 and -2 [18] |
||
|
E7080 [19] |
||
|
BIBF 1120 (vargatef) targets VEGF receptors, FGF receptors, and PDGF receptors, especially in non-small cell lung cancer [20] |
||
|
AZD4547 inhibits FGFR tyrosine kinases 1, 2 and 3 [21] |
||
|
FP-1039 targeting FGFR2 in endometrial carcinoma [22] |
||
|
Platelet derived growth factor (PDGF) |
Activates autocrine and paracrine systems; encourages growth, survival, and motility in malignant, vascular, and stromal cells [23, 24]
|
Imatinib mesylate (PDGFR inhibitor) in a mouse model of cervical carcinogenesis slowed progression of premalignant lesions and impaired growth of invasive carcinoma [25] |
|
Transforming Growth Factor (TGF) |
Induce anchorage‐independent growth of target cells otherwise incapable of such growth [26] |
TGF-β inhibition in hepatocellular carcinoma, colorectal cancer, and glioblastoma multiforme xenograft [27-29] |
|
Galunisertib, (small molecule inhibitor of TGF-βRI) + orafenib, ramucirumab in hepatocellular carcinoma |
||
|
PF-03446962 (a monoclonal antibody against TGF-β) + regorafenib in colorectal carcinoma [30] |
||
|
Tumor Necrosis Factor-α (TNF-α) |
Inflammation; stimulate granulocyte-macrophage-colony stimulating factor (GM-CSF), interleukin-1 (IL-1), and angiogenic factors from other cells; induce endothelial cell differentiation [31] |
Golimumab inhibiting angiogenesis and growth in vivo in metastatic oral squamous cell carcinoma cells [32] |
|
Vascular endothelial growth factor (VEGF) |
Capillary morphogenesis; release of von plasminogen activator (PA), and plasminogen activator receptor (PA-R), Willebrand factor, integrins, and interstitial collagenase; increases vascular permeability and fenestration [33] |
Lung and colon cancer [34, 35] |
Table 2: FDA approved angiogenesis inhibitors as cancer therapeutics (adapted from multiple sources including Yang 2017, Rajabi and Mousa 2017, and Ye 2016) [80-82].
|
Mechanism of Action |
Specific targets |
Generic Name (Brand Name), Year of FDA approval |
Indication(s) |
|
Anti-VEGF antibody |
VEGF-A |
Bevacizumab (Avastin®), 2004 [83] |
Colorectal, non-small-cell lung cancer (NSCLC), glioblastoma multiforme, and epithelial ovarian cancer |
|
VEGF inhibitor (trap mechanism) |
VEGF-A, VEGF-B, PlGF |
Ziv-aflibercept (Zaltrap®), 2012 [84] |
Colorectal cancer |
|
VEGFR antibody |
VEGFR-2 |
Ramucirumab (Cyramza®), 2014 [85, 86] |
Stomach cancer, gastroesophageal junction adenocarcinoma, NSCLC, and hepatocellular carcinoma |
|
Tyrosine kinase inhibitor (TKI) |
VEGFRs, PDGFRs, cKIT |
Axitinib (Inlyta®), 2012 [87] |
Renal cell carcinoma |
|
TKI |
VEGFRs cKIT, DDR2, FGFRs, FLT3, FMS, MUSK, cRAF, PDGFRs, Ret, TAO2 |
Sorafenib (Nexavar®), 2005 [88] |
Renal cell carcinoma and hepatocellular carcinoma |
|
TKI |
VEGFRs ARK5, CaMKIIs, CHK2, cKIT, cRAF, FGFR1, Flt3, FMS, Mer, PDGFR, Ret, TrkA |
Sunitinib (Sutent®), 2006 [89] |
Renal cell carcinoma, gastrointestinal carcinoma and PNETs |
|
TKI |
VEGFRs, cKIT, FMS, FGFR2, FLT3, MLK1, PDGFR |
Pazopanib (Votrient®), 2009 [90, 91] |
Renal cell carcinoma and advanced soft tissue sarcoma |
|
TKI |
VEGFRs PDGFRs, FGFRs, Tie2, DDR2, Trk2A, Eph2A, RAF-1, STK5 |
Regorafenib (Stivarga®), 2012 [92] |
Colorectal cancer, gastrointestinal stromal tumor and hepatocellular carcinoma |
|
TKI |
VEGFRs, cMET, Ret, cKIT, Axl |
Cabozantinib (Cometriq®), 2012 [86, 93] |
Thyroid cancer, renal cell and hepatocellular carcinoma |
|
TKI |
VEGFRs EGFRs, Ret |
Vandetanib (Caprelsa®), 2011 [94] |
Thyroid cancer |
|
bFGF inhibitor |
FGF |
Lenalidomide (Revlimid®) [95, 96] |
Myeloma, mantle cell, follicular and marginal zone lymphoma |
|
bFGF inhibitor |
FGF |
Thalidomide (Synovir, Thalomid®) [95-98] |
Myeloma
|
|
mTOR inhibitor |
mTOR |
Everolimus (Afinitor®) [99] |
Renal cell carcinoma, advanced breast cancer, pancreatic neuroendocrine tumors (PNETs), renal angiomyolipoma, and subependymal giant cell astrocytoma |
|
mTOR inhibitor |
mTOR |
Temsirolimus (Torisel®) [99] |
Renal cell carcinoma |
References
- Adair TH, Montani JP (2010) Overview of Angiogenesis, in Angiogenesis. San Rafael (CA). [Crossref]
- Jain RK (1987) Transport of molecules in the tumor interstitium: a review. Cancer Res 47: 3039-3051. [Crossref]
- Ruoslahti E (2002) Specialization of tumour vasculature. Nat Rev Cancer 2: 83-90. [Crossref]
- Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM (2003) Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol 163: 1801-1815. [Crossref]
- Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 7: 452-464. [Crossref]
- Zhao Y, Adjei AA (2015) Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 20: 660-673. [Crossref]
- Reynolds LP, Grazul Bilska AT, Redmer DA (2000) Angiogenesis in the corpus luteum. Endocrine 12: 1-9. [Crossref]
- Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E et al. (2008) A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 456: 809-813. [Crossref]
- Papetti M, Herman IM (2002) Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol 282: C947- C970. [Crossref]
- Griffioen AW (2008) Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother 57: 1553-1538. [Crossref]
- Lv X, Fang C, Yin R, Qiao B, Shang R et al. (2017) Agrin para-secreted by PDGF-activated human hepatic stellate cells promotes hepatocarcinogenesis in vitro and in vivo. Oncotarget 8: 105340-105355. [Crossref]
- Njah K, Chakraborty S, Qiu B, Arumugam S, Raju A et al. (2019) A Role of Agrin in Maintaining the Stability of Vascular Endothelial Growth Factor Receptor-2 during Tumor Angiogenesis. Cell Rep 28: 949.e7-965.e7. [Crossref]
- Patthy L, Nikolics K (1993) Functions of agrin and agrin-related proteins. Trends Neurosci 16: 76-81. [Crossref]
- Fiedler U, Augustin HG (2006) Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol 27: 552-558. [Crossref]
- Oliner J, Min H, Leal J, Yu D, Rao S et al. (2004) Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 6: 507-516. [Crossref]
- White RR, Shan S, Rusconi CP, Shetty G, Dewhirst MW et al. (2003) Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc Natl Acad Sci U S A 100: 5028-5033. [Crossref]
- Ueda Y, Shimoyama T, Murakami H, Yamamoto N, Yamada Y et al. (2011) Phase I and pharmacokinetic study of TSU-68, a novel multiple receptor tyrosine kinase inhibitor, by twice daily oral administration between meals in patients with advanced solid tumors. Cancer Chemother Pharmacol 67: 1101-1109. [Crossref]
- Dempke WC, Zippel R (2010) Brivanib, a novel dual VEGF-R2/bFGF-R inhibitor. Anticancer Res 30: 4477-4483. [Crossref]
- Yamada K, Yamamoto N, Yamada Y, Nokihara H, Fujiwara Y et al. (2011) Phase I dose-escalation study and biomarker analysis of E7080 in patients with advanced solid tumors. Clin Cancer Res 17: 2528-2537. [Crossref]
- Santos ES, Gomez JE, Raez LE (2012) Targeting angiogenesis from multiple pathways simultaneously: BIBF 1120, an investigational novel triple angiokinase inhibitor. Invest New Drugs 30: 1261-1269. [Crossref]
- Gavine PR, Mooney L, Kilgour E, Thomas A, Al Kadhimi K et al. (2011) Abstract 3568: Characterization of AZD4547: An orally bioavailable, potent and selective inhibitor of FGFR tyrosine kinases 1, 2 and 3. Am Assoc Cancer Res.
- Harding TC, Palencia S, Long L, Finer J, Keer HN et al. (2010) Abstract 2597: Preclinical efficacy of FP-1039 (FGFR1: Fc) in endometrial carcinoma models with activating mutations in FGFR2. Am Assoc Cancer Res.
- Tejada ML, Yu L, Dong J, Jung K, Meng G et al. (2006) Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin Cancer Res 12: 2676-2688. [Crossref]
- Cao Y (2013) Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med 19: 460-473. [Crossref]
- Lev DC, Kim SJ, Onn A, Stone V, Nam DH et al. (2005) Inhibition of platelet-derived growth factor receptor signaling restricts the growth of human breast cancer in the bone of nude mice. Clin Cancer Res 11: 306-314. [Crossref]
- Lawrence DA (1985) Transforming growth factors--an overview. Biol Cell 53: 93-98. [Crossref]
- Mazzocca A, et al. (2009) Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology 50: 1140-1151. [Crossref]
- Akbari A, Amanpour S, Muhammadnejad S, Ghahremani MH, Ghaffari SH et al. (2014) Evaluation of antitumor activity of a TGF-beta receptor I inhibitor (SD-208) on human colon adenocarcinoma. Daru 22: 47. [Crossref]
- Zhang M, Kleber S, Röhrich M, Timke C, Han N et al. (2011) Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res 71: 7155-7167. [Crossref]
- Comunanza V, Bussolino F (2017) Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front Cell Dev Biol 5: 101. [Crossref]
- Klagsbrun M, D'Amore PA (1991) Regulators of angiogenesis. Annu Rev Physiol 53: 217-239. [Crossref]
- Lai KC, Liu C, Lin T, Mar A, Wang H et al. (2016) Blocking TNF-alpha inhibits angiogenesis and growth of IFIT2-depleted metastatic oral squamous cell carcinoma cells. Cancer Lett 370: 207-215. [Crossref]
- Ucuzian AA, Gassman AA, East AT, Greisler HP (2010) Molecular mediators of angiogenesis. J Burn Care Res 31: 158-175. [Crossref]
- Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438: 932-936. [Crossref]
- Hurwitz H (2004) Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin Colorectal Cancer 4: S62-S68. [Crossref]
- Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1: 27-31. [Crossref]
- Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G (1996) Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Res 56: 1111-1117. [Crossref]
- Dirkx AE, Oude Egbrink MGA, Wagstaff J, Griffioen AW (2006) Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol 80: 1183-1196. [Crossref]
- Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J et al. (1996) Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res 56: 4625-4629. [Crossref]
- Ferrara N, Adamis A (2016) Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov 15: 385-403. [Crossref]
- Nishida N, Yano H, Nishida T, Kamura T, Kojiro M (2006) Angiogenesis in cancer. Vasc Health Risk Manag 2: 213-219. [Crossref]
- Alitalo K, Tammela T, Petrova TV (2005) Lymphangiogenesis in development and human disease. Nature 438: 946-953. [Crossref]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N (1992) Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem 267: 26031-26037. [Crossref]
- Park JE, Chen HH, Winer J, Houck KA, Ferrara N (1994) Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 269: 25646-25654. [Crossref]
- Wang S, Li X, Parra M, Verdin E, Bassel Duby R et al. (2008) Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci U S A 105: 7738-7743. [Crossref]
- Takahashi T, Yamaguchi S, Chida K, Shibuya M (2001) A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20: 2768-2778. [Crossref]
- Takahashi T, Ueno H, Shibuya M (1999) VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 18: 2221-2230. [Crossref]
- Karaman S, Leppanen VM, Alitalo K (2018) Vascular endothelial growth factor signaling in development and disease. Development 145: dev151019. [Crossref]
- Tirapu I, Huarte E, Guiducci C, Arina A, Zaratiegui M et al. (2006) Low surface expression of B7-1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res 66: 2442-2450. [Crossref]
- Rodríguez JA (2017) HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol Lett 14: 4415-4427. [Crossref]
- Chaudhary B, Elkord E (2016) Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines (Basel) 4: 28. [Crossref]
- Zou W (2005) Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5: 263-274. [Crossref]
- Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH (1995) Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med 181: 811-816. [Crossref]
- Kazemi S, Wenzel D, Kolossov E, Lenka N, Raible A et al. (2002) Differential role of bFGF and VEGF for vasculogenesis. Cell Physiol Biochem 12: 55-62. [Crossref]
- Teicher BA, Holden SA, Ara G, Korbut T, Menon K (1996) Comparison of several antiangiogenic regimens alone and with cytotoxic therapies in the Lewis lung carcinoma. Cancer Chemother Pharmacol 38: 169-177. [Crossref]
- Griffioen AW, Damen CA, Mayo KH, Barendsz Janson AF, Martinotti S et al. (1999) Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int J Cancer 80: 315-319. [Crossref]
- Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P et al. (2015) The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 26: 507-520. [Crossref]
- Kuijper H, Gallardo Torres HI, van der Linden JA, Lammers JW, Sixma JJ et al. (1996) Platelet-dependent primary hemostasis promotes selectin- and integrin-mediated neutrophil adhesion to damaged endothelium under flow conditions. Blood 87: 3271-3281. [Crossref]
- Semple JW, Italiano JE Jr, Freedman J (2011) Platelets and the immune continuum. Nat Rev Immunol 11: 264-274. [Crossref]
- Lertkiatmongkol P, Liao D, Mei H, Hu Y, Newman PJ (2016) Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr Opin Hematol 23: 253-259. [Crossref]
- Melder RJ, Koenig GC, Witwer BP, Safabakhsh N, Munn LL et al. (1996) During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat Med 2: 992-997. [Crossref]
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM et al. (1996) Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 2: 1096-1103. [Crossref]
- Laxmanan S, Robertson SW, Wang E, Lau JS, Briscoe DM et al. (2005) Vascular endothelial growth factor impairs the functional ability of dendritic cells through Id pathways. Biochem Biophys Res Commun 334: 193-198. [Crossref]
- Yang DH, Park JS, Jin CJ, Kang HK, Nam JH et al. (2009) The dysfunction and abnormal signaling pathway of dendritic cells loaded by tumor antigen can be overcome by neutralizing VEGF in multiple myeloma. Leuk Res 33: 665-670. [Crossref]
- Puig Kröger A, Relloso M, Fernández Capetillo O, Zubiaga A, Silva A et al. (2001) Extracellular signal-regulated protein kinase signaling pathway negatively regulates the phenotypic and functional maturation of monocyte-derived human dendritic cells. Blood 98: 2175-2182. [Crossref]
- Loscher CE, Draper E, Leavy O, Kelleher D, Mills KHG et al. (2005) Conjugated linoleic acid suppresses NF-kappa B activation and IL-12 production in dendritic cells through ERK-mediated IL-10 induction. J Immunol 175: 4990-4998. [Crossref]
- Heine A, Erika Held SA, Daecke SN, Riethausen K, Kotthoff P et al. (2015) The VEGF-Receptor Inhibitor Axitinib Impairs Dendritic Cell Phenotype and Function. PLoS One 10: e0128897. [Crossref]
- Heine A, Holderried TA, Brossart P (2009) Immunotherapy in renal cell carcinoma. Immunotherapy 1: 97-107. [Crossref]
- Bose A, Lowe DB, Rao A, Storkus WJ (2012) Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res 22: 236-243. [Crossref]
- Mimura K, Kono K, Takahashi A, Kawaguchi Y, Fujii H (2007) Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother 56: 761-770. [Crossref]
- Young MR, Kolesiak K, Wright MA, Gabrilovich DI (1999) Chemoattraction of femoral CD34+ progenitor cells by tumor-derived vascular endothelial cell growth factor. Clin Exp Metastasis 17: 881-888. [Crossref]
- Riboldi E, Musso T, Moroni E, Urbinati C, Bernasconi S et al. (2005) Cutting edge: proangiogenic properties of alternatively activated dendritic cells. J Immunol 175: 2788-2792. [Crossref]
- Fernandez Pujol B, Lucibello FC, Zuzarte M, Lütjens P, Müller R et al. (2001) Dendritic cells derived from peripheral monocytes express endothelial markers and in the presence of angiogenic growth factors differentiate into endothelial-like cells. Eur J Cell Biol 80: 99-110. [Crossref]
- Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M et al. (2015) VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 212: 139-148. [Crossref]
- Vitale G, Dicitore A, Gentilini D, Cavagnini F (2010) Immunomodulatory effects of VEGF: Clinical implications of VEGF-targeted therapy in human cancer. Cancer Biol Ther 9: 694-698. [Crossref]
- Yang J, Yan J, Liu B (2018) Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front Immunol 9: 978. [Crossref]
- El Kenawi AE, El Remessy AB (2013) Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol 170: 712-729. [Crossref]
- Kerbel R, Folkman J (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2: 727-739. [Crossref]
- Clarke S, Sharma R (2006) Angiogenesis inhibitors in cancer - mechanisms of action. Aust Prescr 29: 9-12.
- Yang WH, Xu J, Mu JB, Xi J (2017) Revision of the concept of anti-angiogenesis and its applications in tumor treatment. Chronic Dis Transl Med 3: 33-40. [Crossref]
- Rajabi M, Mousa SA (2017) The Role of Angiogenesis in Cancer Treatment. Biomedicines 5: 34. [Crossref]
- Ye W (2016) The Complexity of Translating Anti-angiogenesis Therapy from Basic Science to the Clinic. Dev Cell 37: 114-125. [Crossref]
- Monk BJ, Randall LM, Grisham RN (2019) The Evolving Landscape of Chemotherapy in Newly Diagnosed Advanced Epithelial Ovarian Cancer. Am Soc Clin Oncol Educ Book 39: e141-e151. [Crossref]
- Muro K, Salinardi T, Singh AR, Macarulla T (2020) Safety of Aflibercept in Metastatic Colorectal Cancer: A Literature Review and Expert Perspective on Clinical and Real-World Data. Cancers (Basel) 12: 844. [Crossref]
- Larkins E, Scepura B, Blumenthal GM, Bloomquist E, Tang S et al. (2015) U.S. Food and Drug Administration Approval Summary: Ramucirumab for the Treatment of Metastatic Non-Small Cell Lung Cancer Following Disease Progression On or After Platinum-Based Chemotherapy. Oncologist 20: 1320-1325. [Crossref]
- Mahipal A, Tella SH, Kommalapati A, Lim A, Kim R (2019) Immunotherapy in Hepatocellular Carcinoma: Is There a Light at the End of the Tunnel? Cancers (Basel) 11: 1078. [Crossref]
- Bellesoeur A, Carton E, Alexandre J, Goldwasser F, Huillard O (2017) Axitinib in the treatment of renal cell carcinoma: design, development, and place in therapy. Drug Des Devel Ther 11: 2801-2811. [Crossref]
- Raoul JL, Kudo M, Finn RS, Edeline J, Reig M et al. (2018) Systemic therapy for intermediate and advanced hepatocellular carcinoma: Sorafenib and beyond. Cancer Treat Rev 68: 16-24. [Crossref]
- Motzer RJ, Escudier B, Gannon A, Figlin RA (2017) Sunitinib: Ten Years of Successful Clinical Use and Study in Advanced Renal Cell Carcinoma. Oncologist 22: 41-52. [Crossref]
- Gill DM, Agarwal N, Vaishampayan U (2017) Evolving Treatment Paradigm in Metastatic Renal Cell Carcinoma. Am Soc Clin Oncol Educ Book 37: 319-329. [Crossref]
- Dembla V, Groisberg R, Hess K, Fu S, Wheler J et al. (2017) Outcomes of patients with sarcoma enrolled in clinical trials of pazopanib combined with histone deacetylase, mTOR, Her2, or MEK inhibitors. Sci Rep 7: 15963. [Crossref]
- Ettrich TJ, Seufferlein T (2018) Regorafenib. Recent Results Cancer Res 211: 45-56. [Crossref]
- Abdelaziz A, Vaishampayan U (2017) Cabozantinib for Renal Cell Carcinoma: Current and Future Paradigms. Curr Treat Options Oncol 18: 18. [Crossref]
- Fallahi P, Ferrari SM, Elia G, Ragusa F, Paparo SR et al. (2019) Evaluating vandetanib in the treatment of medullary thyroid cancer: patient-reported outcomes. Cancer Manag Res 11: 7893-7907. [Crossref]
- Fuchs O (2019) Treatment of Lymphoid and Myeloid Malignancies by Immunomodulatory Drugs. Cardiovasc Hematol Disord Drug Targets 19: 51-78. [Crossref]
- Yamshon S, Ruan J (2019) IMiDs New and Old. Curr Hematol Malig Rep 14: 414-425. [Crossref]
- Mujagic H, Chabner BA, Mujagic Z (2002) Mechanisms of action and potential therapeutic uses of thalidomide. Croat Med J 43: 274-285. [Crossref]
- Zhou S, Wang F, Hsieh TC, Wu JM, Wu E (2013) Thalidomide-a notorious sedative to a wonder anticancer drug. Curr Med Chem 20: 4102-4108. [Crossref]
- Janku F, Yap TA, Meric Bernstam F (2018) Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 15: 273-291. [Crossref]
- Baka S, Clamp AR, Jayson GC (2006) A review of the latest clinical compounds to inhibit VEGF in pathological angiogenesis. Expert Opin Ther Targets 10: 867-876. [Crossref]
- Meadows KL, Hurwitz HI (2012) Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med 2: a006577. [Crossref]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J et al. (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335-2342. [Crossref]
- Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP et al. (2007) Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 25: 1539-1544. [Crossref]
- Saltz LB, Clarke S, Díaz Rubio E, Scheithauer W, Figer A et al. (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26: 2013-2019. [Crossref]
- Galustian C, Dalgleish A (2009) Lenalidomide: a novel anticancer drug with multiple modalities. Expert Opin Pharmacother 10: 125-133. [Crossref]
- Highleyman L (1998) FDA approves fomivirsen, famciclovir, and Thalidomide. Food and Drug Administration. BETA 5. [Crossref]
- Abdel Qadir H, Ethier JL, Lee DS, Thavendiranathan P, Amir E (2017) Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat Rev 53: 120-127. [Crossref]
- Izzedine H, Rixe O, Billemont B, Baumelou A, Deray G (2007) Angiogenesis inhibitor therapies: focus on kidney toxicity and hypertension. Am J Kidney Dis 50: 203-218. [Crossref]
- Kappers MH, van Esch JHM, Sleijfer S, Danser AHJ, van den Meiracker AH (2009) Cardiovascular and renal toxicity during angiogenesis inhibition: clinical and mechanistic aspects. J Hypertens 27: 2297-2309. [Crossref]
- Puthillath A, Patel A, Fakih MG (2009) Targeted therapies in the management of colorectal carcinoma: role of bevacizumab. Onco Targets Ther 2: 1-15. [Crossref]
- Ellis LM (2006) Mechanisms of action of bevacizumab as a component of therapy for metastatic colorectal cancer. Semin Oncol 33: S1-S7. [Crossref]
- Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R et al. (2017) Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 9: eaak9679. [Crossref]
- Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ et al. (2004) Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res 64: 3731-3736. [Crossref]
- Li X, Wu M, Pan L, Shi J (2016) Tumor vascular-targeted co-delivery of anti-angiogenesis and chemotherapeutic agents by mesoporous silica nanoparticle-based drug delivery system for synergetic therapy of tumor. Int J Nanomedicine 11: 93-105. [Crossref]
- Dings RP, Loren M, Heun H, McNiel E, Griffioen AW et al. (2007) Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clin Cancer Res 13: 3395-3402. [Crossref]
- Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8: 425-437. [Crossref]
- Craig DJ (2020) The Abscopal Effect of Radiation Therapy. Future Oncol.
- Barve M, Kuhn J, Lamont J, Beitsch P, Manning L et al. (2016) Follow-up of bi-shRNA furin / GM-CSF Engineered Autologous Tumor Cell (EATC) Immunotherapy Vigil® in patients with advanced melanoma. Biomed Genet Genom 1.
- Ghisoli M, Barve M, Mennel R, Lenarsky C, Horvath S et al. (2016) Three-year Follow up of GMCSF/bi-shRNA(furin) DNA-transfected Autologous Tumor Immunotherapy (Vigil) in Metastatic Advanced Ewing's Sarcoma. Mol Ther 24: 1478-1483. [Crossref]
- Ghisoli M, Barve M, Schneider R, Mennel R, Lenarsky C et al. (2015) Pilot Trial of FANG Immunotherapy in Ewing's Sarcoma. Mol Ther 23: 1103-1109. [Crossref]
- Oh J, Barve M, Matthews CM, Koon EC, Heffernan TP et al. (2016) Phase II study of Vigil® DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol Oncol 143: 504-510. [Crossref]
- Oh J, Barve M, Senzer N, Aaron P, Manning L et al. (2020) Long-term follow-up of Phase 2A trial results involving advanced ovarian cancer patients treated with Vigil® in frontline maintenance. Gynecol Oncol Rep 34: 100648. [Crossref]
- Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P et al. (2012) Phase I trial of "bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell" vaccine (FANG) in advanced cancer. Mol Ther 20: 679-686. [Crossref]
- Senzer N (2013) Long Term Follow Up: Phase I Trial of “bi-shRNA furin/GMCSF DNA/Autologous Tumor Cell” Immunotherapy (FANG™) in Advanced Cancer. J Vaccines Vaccination 4: 209.
- Herron J, Smith N, Stanbery L, Aaron P, Manning L et al. (2020) Vigil: Personalized Immunotherapy Generating Systemic Cytotoxic T cell Response. Cancer Sci Res 3: 1-4.
- Rocconi RP, Grosen EA, Ghamande SA, Chan JK, Barve MA et al. (2020) Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol 21: 1661-1672. [Crossref]
- McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R et al. (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351: 1463-1469. [Crossref]
- Amin A, Plimack ER, Ernstoff MS, Lewis LD, Bauer TM et al. (2018) Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: the CheckMate 016 study. J Immunother Cancer 6: 109. [Crossref]
- Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott DF et al. (2018) Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol 19: 405-415. [Crossref]
- Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R et al. (2019) Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 380: 1116-1127. [Crossref]
- Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L et al. (2019) Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 380: 1103-1115. [Crossref]
- McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI et al. (2018) Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 24: 749-757. [Crossref]
- Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K et al. (2016) Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 7: 12624. [Crossref]
- Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D et al. (2018) Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 378: 2288-2301. [Crossref]
- Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F et al. (2017) Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389: 255-265. [Crossref]
- Hegde S, Wallin JJ, Mancao C (2018) Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol 52: 117-124. [Crossref]
- Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F et al. (2019) Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med 7: 387-401. [Crossref]
- Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S et al. (2020) Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol 21: 808-820. [Crossref]
- Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M et al. (2020) Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 382: 1894-1905. [Crossref]