Journals
Ion channels involved in spontaneous pain
A B S T R A C T
Pain is a distressing feeling that is often induced by damaging stimuli. The nerve injuries cause various molecular changes in nociceptive primary afferent neurons that cause spontaneous pain. Furthermore, the nerve trunk injury induces ectopic discharge, resulting in spontaneous pain. To date, accumulating evidence suggests that ion channels which are responsible for neuronal excitability play key roles in generation of spontaneous pain. It is believed that voltage-gated Na+ channels (VGSCs) and Ca2+ channels (VGCCs) are the primary membrane proteins for causing spontaneous pain. However, it has been evident that various ion channels including transient receptor potential (TRP) channels, hyperpolarization-activated cyclic nucleotide–gated (HCN) channels and acid-sensing ion channels (ASICs) are associated with generation of spontaneous pain. In the present review, I will describe the current knowledge on ion channels related to spontaneous pain.
K E Y W O R D S
ion channel, spontaneous pain, neuron
I N T R O D U C T I O N
The sensation of pain is generally caused by activities of Aδ- and C-primary afferent nociceptive neurons. In the absence of stimuli, these nociceptive neurons are silent at rest, but become active in respond to noxious stimuli. However, after injury to peripheral nerves, nociceptive neurons become unusually sensitive and generate abnormal spontaneous activity [1]. The pain sensation is thus generated by spontaneous activity in injured and nearby nociceptive afferent fibers. Spontaneous pain is described in the context of chronic neuropathic and chronic inflammatory pain conditions. The mechanisms underlying the spontaneous pain are poorly understood. There is growing evidence that spontaneous action potential firings in neurons that transmit pain signals from the injury site to the spinal cord is responsible for spontaneous pain [2]. Although it remains largely unknown how the spontaneous firing is caused, neuronal excitability that is regulated by ion channels would play the essential role. Several studies in animals and humans demonstrated that the increased expression of Na+ channels is involved the occurrence of spontaneous pain [1]. Other studies also suggested that modulation of K+ and Ca2+ channels plays key roles in spontaneous firings [1]. Therefore, it is highly possible that spontaneous pain is caused by dysfunction of ion channels. In this review, I will describe evidence that several ion channels are responsible for spontaneous pain (Figure 1).
Na+ Channel
Na+ channels are composed of transmembrane proteins that have voltage-gated pore, where Na+ ions are selectively transported across the membrane. The activity of voltage-gated Na+ channels (VGSCs) is critical for the excitability and conductivity in neurons [3]. The VGSCs consist of large pore-forming α subunits (260 kDa), that may be associated with auxiliary β subunits [4]. Until now, it is known that nine pore-forming α subunits (Nav1.1-Nav1.9) are present and these channels are broadly distributed in the peripheral and central nervous system (PNS and CNS. respectively) [5, 6]. Among these voltage-gated Na+ channels, Nav1.1, Nav1.2 and Nav1.3 are predominantly expressed in the CNS, whereas Nav1.6, Nav1.7, Nav1.8 and Nav1.9 are mainly expressed in the PNS [3]. In sensory neurons, the VGSCs are critical determinants of the electrical excitability and involved in pain perception by regulating afferent impulse discharges. The composition of voltage-gated Na+ channels may undergo significant changes upon nerve injury.
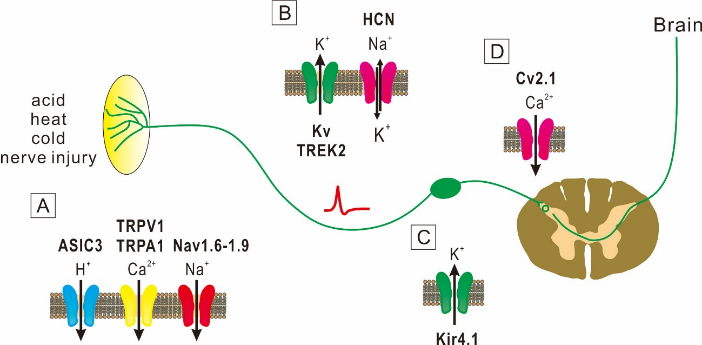
A: At peripheral nerve endings of DRG, ASIC3, TRPA1, TRPV1 and Nav1.6-1.9 channels mediate spontaneous pain.
B: Kv channel α subunit genes, TREK2 and HCN channels, which are involved in action potential conduction, subserve spontaneous pain.
C: Kir4.1 channels that are expressed in glial cells are involved in spontaneous pain.
D: Cv2.1 channels that modulate transmitter release are involved in spontaneous pain.
Figure 1: Key ion channels involved in spontaneous pain.
After axonal damage, Na+ channels accumulate at the sites of sprouting [7]. Pharmacological experiments revealed that VGSCs play an important role in spontaneous electrogenesis in neurons [8]. The accumulation of VGSCs at sites of ectopic nerve impulse generation could be associated with the lowing of the threshold for action potential and the subsequent hyperactivity [3]. Among several VGSCs, it has been demonstrated that Nav1.6-1.9 are involved in the generation of ectopic (spontaneous) action potentials following after axonal damage, resulting in the spontaneous pain [9]. Nav1.7 channels are predominantly present in the dorsal root ganglion (DRG) and produce a rapidly activating and inactivating current which is sensitive to tetrodotoxin [9]. Nav1.7 channels contribute to the peripheral nociceptive sensory neuron excitability and carry nociceptive signals into the spinal cord, presumably through facilitation of action potential propagation [9]. Several clinical genetic researches have demonstrated that deficiency of function of Nav1.7 channels (encoded by the SCN9A gene) caused complete inability to perceive pain [10]. In addition, a gain-of-function Nav1.7 mutation caused or contributed to chronic spontaneous pain [11, 12]. Nav1.8 channels produce slowly inactivating currents which are preferentially expressed in the trigeminal ganglion and DRG [9]. Several lines of evidence have supported that Nav1.8 channels play an essential role in pain perception. Nav1.8 channels contribute substantially to the Na+ currents underlying the rising phase of action potential in the C-type DRG neurons [13]. It has also been reported that sensory neurons expressing Nav1.8 are pivotal for responses to cold and noxious mechanical pressure but not acute noxious heat [14]. Furthermore, Nav1.8 channels were suggested to be involved in inflammatory pain but not for neuropathic pain [14]. A mutation study of SCN10A that is a gene encoding Nav1.8 has shown the relationship between Nav1.8 channels and human pain [15]. Nav1.9 channels are mainly present in small-sized neurons of DRG and trigeminal ganglion [9]. Nav1.9 channels show slower activation kinetics than that of Nav1.8 channels, and consequently are not responsible for the action potential upswing [16]. Furthermore, Nav1.9 channels show slow inactivation kinetics over a wide voltage range, rendering it to generate a component of persistent Na+ currents [16]. Therefore, Nav1.9 channels could play a critical role in determining the excitability of nociceptive neurons by modulation of the resting membrane potential and responses to subthreshold stimuli. A direct role of Nav1.9 channels in the spontaneous pain responses and persistent hypersensitivity to heat following peripheral inflammation and nerve injury has been demonstrated in a study using mutations of SCN11A, the gene that encodes Nav1.9 [17]. It has also been demonstrated that using Nav1.9 knockout mice and Nav1.9 knockdown rats, Nav1.9 channels play a critical role in generation of hypersensitivity to mechanical and thermal stimuli both in chronic and subacute inflammatory pain conditions [18]. In comparison with the functional roles of Nav1.7-1.9 channels, little information is available for the functional role of Nav1.6 channels in peripheral sensory neurons. However, it has been shown that an injection of a Nav1.6 activator induced mechanical allodynia and spontaneous pain, and increased K+-channel blocker 4-aminopyridine-induced cold allodynia [19]. These findings suggest a critical role of Nav1.6 channels in multiple pain pathways from peripheral nociceptors. Taken together, it is strongly suggested that Nav channels are key players in the establishment of spontaneous pain.
K+ Channel
K+ channels are critical determinants of membrane excitability in the CNS. A large number of studies revealed an involvement of K+ channels in processing of nociceptive stimuli. An application of K+ channel activators on the axon terminals or cell bodies of DRG neurons decreases the membrane excitability while K+ channel inhibitors increase spike firings [20]. Thus, it is possible that in chronic pain state, inhibition of K+ channels cause the spontaneous activities in neurons. K+ channels are grouped into three families based on structural properties [21]. The first family is the voltage-gated K+ channels such as the transient voltage-dependent and delayed rectifier K+ channels and Ca2+-dependent K+ channels [22, 23]. The second family is the inwardly-rectifying K+ channels such as ATP-sensitive K+ channels [24]. The third family is the two-pore-domain K+ (leak K+) channels [21, 25].
Voltage-gated potassium (Kv) channels are composed of ion-conducting subunits and auxiliary cytoplasmic subunits. The Kv channels subserve important roles in setting the resting membrane potentials, mediating the repolarization of action potential and controlling the subthreshold membrane potential oscillations [22]. The Kv superfamily is known to be composed of 40 human genes [26]. These K+ channels are further grouped into 12 classes. To date, many Kv channels are known to be responsible for the inflammatory and neuropathic pain [27]. Several Kv channels are reported to be involved in the spontaneous pain [27]. Following chronic constriction injury in rats, the expression of Kv channel gene was decreased in the DRG neurons and ectopic spontaneous discharge was generated at primary sensory neurons [28]. The downregulation of Kv9.1 expression after nerve injury triggered mechanical allodynia as well as spontaneous and evoked hyperexcitability [29]. Thus, the downregulation of the Kv channel exerts an important role in spontaneous pain.
Inwardly rectifying K+ channels (Kir) are the two-transmembrane domain K+ channels. To date, seven subfamilies have been identified in mammalian cells, and they are divided into four functional groups [30]. Kir2.1, Kir2.2 and Kir2.3 are constitutively active while Kir3.1, Kir3.2, Kir3.3 and Kir3.4 are regulated by G protein-coupled receptors. Kir6.2 and Kir6.2 are ATP-sensitive K+ channel families, which are tightly associated with cellular metabolism. Kir1, Kir4, Kir5 and Kir7 are known as K+ transport channels [30]. Kir channels are widely expressed in the CNS and PNS [31]. Similar to the Kv channels, accumulating evidence suggests that Kir are responsible for neuropathic and inflammatory pain [27].
It is well established that the Kir channels are expressed in supporting cells such as glia. The glia expresses diverse Kir channels, whose major function is to establish the high K+ selectivity of the cell membrane of glial cells and to set the resting membrane potential. Among several Kir channels, Kir4.1 has a major regulatory role in glia [31]. For example, knockdown of Kir4.1 expression in trigeminal ganglion neurons caused membrane hyperexcitability [32]. In Kir4.1 knockout mice, inward currents were almost completely absent together with the depolarized resting membrane potential [33]. Furthermore, it has been shown that Kir4.1 gene silencing by RNA interference resulted in the pain-like behavior in rats without nerve injury [34]. In addition, following chronic constriction injury of the infraorbital nerve, the expression level of Kir channels was decreased in the trigeminal ganglion [34]. These observations suggest that spontaneous pain is brought about by the decreased expression of Kir4.1 in glial cells. Thus, targeting Kir4.1 channel could be a useful treatment for spontaneous pain.
The two-pore-domain K+ (leak K+) channels are open at rest and are the main contributors to resting membrane potential in neurons [35]. The leak K+ channels contain four transmembrane and two pore domains [21, 25]. In mammals, 15 gene families are known, and they are grouped into six subfamilies: 1) TWIK, tandem of pore domains in a weak inward rectifying K+ channel, 2) TREK, TWIK-related K+ channel, 3) TASK, TWIK-related acid-sensitive K+ channel, 4) THIK, TWIK-related halothane-inhibited K+ channel, 5) TRESK, TWIK-related spinal cord K+ channel, 6) TALK, TWIK-related alkali-activated sensitive K+ channel [25, 35]. The TWIK group includes TWIK1, TWIK2 and KCNK7. The THIK group includes THIK1 and THIK2. The TREK group includes TREK1, TREK2 and TRAAK. The TALK group includes TASK2, TALK1 and TALK2/TASK4. The TASK group includes TASK1, TASK3 and TASK5. The TRESK group includes TRESK1. Among these channels, it has been reported that TREK2 channels are involved in spontaneous pain. In DRG neurons, TREK2 channels are a pivotal determinant of the resting membrane potential [36]. It has also been demonstrated that TREK2 channels were selectively expressed in C-fiber nociceptors and inhibit spontaneous pain [36]. The knockdown of TREK2 with siRNA induced spontaneous pain behavior [36]. These observations suggest that TREK2 channels hyperpolarize C-fiber nociceptive neurons and inhibits spontaneous pain.
Voltage-gatedCa2+channels(VGCCs)
VGCCs play critical roles in diverse physiological functions. To date, ten members of the VGCC family are described in mammals (Cav1.1, Cav1.2, Cav1.3, Cav1.4, Cav2.1, Cav2.2, Cav2.3, Cav3.1, Cav3.2 and Cav3.3), and these channels show differential roles in intracellular signal transduction [37]. Cav1.1, Cav1.2, Cav1.3 and Cav1.4 are grouped into L-type VGCCs, Cav2.1 is grouped into N-type VGCCs, Cav2.2 is grouped into P/Q-type VGCCs, Cav2.3 is grouped into R-type VGCCs, and Cav3.1, Cav3.2 and Cav3.3 are grouped into T-type VGCCs [37]. The family of Cav1 channels is associated with synaptic transmission and integration of synaptic inputs in neurons. The family of Cav2 channels is involved in initiation of synaptic transmission. The family of Cav3 channels is involved in repetitive action potential firings in thalamic neurons [38]. Among ten VGCCs, N- and P-type VGCCs are predominantly expressed in neuronal tissue in the brain, and Ca2+ influx through these channels is essential for depolarization-induced transmitter release [39-41]. Antagonists for N- and P-type VGCCs showed antinociceptive effects in animal models of inflammation, suggesting a role of Ca2+ influx into sensory neurons in the processing of nociceptive inputs that occurs at the spinal level [42]. Blockade of N-type VGCCs has been shown to reduce secondary heat hyperalgesia and spontaneous pain-related behaviors associated with acute joint inflammation [43]. Also, it has been shown that blockade of N-type VGCCs was effective on nociceptive responses in spinal dorsal horn neurons following nerve injury [44]. Thus, N-type VGCCs are important for generation of spontaneous pain.
Transient Receptor Potential (TRP) Channels
Transient receptor potential vanilloid 1 (TRPV1), known as the capsaicin receptor, is a ligand-gated ion channel. TRPV1 channels are activated by multiple pain stimuli such as acid, heat, capsaicin, protons, lipids and spider toxins. Furthermore, the activity of TRPV1 channels is enhanced by a number of inflammatory mediators including bradykinin, prostaglandins, nerve growth factor and ATP [45]. TRPV1 channels are predominately localized in small C-type fibers which mediate pain, and also present in lamina I and II of the spinal dorsal horn [46, 47]. Furthermore, TRPV1 channels are localized at the supraspinal level and contribute to descending modulation of nociceptive transmission [48]. Both the gene deletion and pharmacological studies have shown that TRPV1 channels have central roles in inflammatory and neuropathic pain [49]. A previous study demonstrated that a potent and selective TRPV1 antagonist, ABT-102, was effective in suppression of nociceptive pain in rodent various pain models: ABT-102 depressed spontaneous pain behaviors [50]. Therefore, antagonist for TRPV1 channels could be effective for treatment of spontaneous pain.
Transient receptor potential ankyrin 1 (TRPA1), known as a noxious cold-activated ion channel, is nonselective cation channels that are mainly expressed on nociceptive primary afferent sensory neurons [51]. At peripheral terminals of nociceptive sensory neurons, TRPA1 channels contribute to transmitting harmful stimuli, whereas at central terminals in the spinal dorsal horn, these channels regulate excitatory synaptic transmission to interneurons in the spinal cord. Previous findings suggested the involvement of TRPA1 in chronic and acute nociceptive processes to cold stimuli [52]. It was reported that TRPA1 contributed to spontaneous pain-like behaviors caused by endothelin-1 in mice [53]. It has also been demonstrated that TRPA1 was involved in postoperative pain in the rat [54]. In their study, intrathecal treatment of a TRPA1 antagonist attenuated hypersensitivity but not spontaneous pain-like behavior, suggesting that TRPA1 channels located in the skin are involved in postoperative pain evoked by noxious mechanical stimulus while TRPA1 channels in the spinal cord contribute mainly to postoperative pain caused by innocuous mechanical stimulus. Thus, it is likely that peripheral TRPA1 channels play essential roles in spontaneous pain-like behaviors.
Acid-Sensing Ion Channels (ASICs)
ASICs are H+-gated cationic channels, and ASIC proteins are a subfamily of degenerin (DEG)/epithelial sodium channel (ENaC) superfamily of non-voltage-gated ion channels [55, 56]. In mice and rats, at least six ASIC isoforms (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 and ASIC4) encoded by four separate genes (ACCN1, ACCN2, ACCN3 and ACCN4) have been described [55]. Among six isoforms, ASIC1a, 1b, 2a, 2b, and 3 are present in peripheral sensory neurons while ASIC1a, 2a, and 2b are expressed in the CNS [55]. The ASIC subunits can form functional homomultimers as well as heteromultimers. Although the exact subunit combinations of functional native ASICs have not been identified, the composition of ASIC subunits determines the pH-sensitivity, ionic selectivity and kinetics of activation and desensitization. For example, the desensitization rate of the transient current in ASIC1a/ASIC2a heteromeric channels is faster than that of ASIC1a or ASIC2a homomeric channels [57]. The pH of halfmaximal activation of ASIC homomeric channels are described: 5.8–6.8 for ASIC1a, 6.1–6.2 for ASIC1b, 4.5–4.9 for ASIC2a, and 6.4–6.6 for ASIC3 [56]. Thus, the extracellular acidification activates ASICs under the physiological and pathophysiological states. Several studies demonstrated that inflammation and nerve injury induce acidosis, and acidic low pH can cause pain [58, 59]. This suggests that ASICs have a critical role in nociception.
A previous study demonstrated that ASIC3 display a biphasic current in response to acidosis in extracellular pH when these channels were expressed in heterologous cells [60]. In addition to a transient inward current that was rapidly inactivated, ASIC3 showed a non-desensitizing sustained current that lasts as long as the acidic extracellular pH is maintained [61]. ASIC3 are expressed predominantly in DRG neurons including large-diameter mechanoreceptors and C-fiber nociceptors [62]. Therefore, it is conceivable that ASIC3 exert a critical role in nociception by acting as an indicator of tissue acidosis. It has previously been reported that after injection of acetic acid, spontaneous pain behaviors were significantly increased in neuropathic rats induced by the spinal nerve ligation (SNL) of the L5 spinal nerves, compared to the sham-treated group [63]. Consistent with this behavioral change, the immunoreactivity of ASIC3 was significantly upregulated in the nociceptive DRG neurons. These data indicate that ASIC3 are related to spontaneous pain in the L5 SNL rat model. Although it is thought that ASIC3 are involved in acute pain states, it also contributes to spontaneous pain. The antagonists for ASICs may provide a new option for suppressing spontaneous pain.
Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels
HCN channels that generate hyperpolarization-activated currents, comprise four isoforms: HCN1, HCN2, HCN3 and HCN4 [64]. HCN channels are activated at voltages near the typical resting membrane potential and are primarily permeable to Na+ and K+. HCN channels are widely expressed in the heart and in the CNS and PNS [64]. Among four HCN isoforms, HCN1, 2 and 4 channels are responsible for the generation of neuronal activity, whereas the functional role of HCN3 channels remains largely unclear. Several studies have showed that the hyperpolarization-activated currents regulate the neuronal membrane excitability by regulating basic membrane properties in both the physiological and pathological states [64]. HCN channels are known as the pacemaker channels since they help to generate rhythmic activities of cells both in the brain and heart [64]. Furthermore, it has been reported that HCN channels contribute to generation of neuropathic pain, and thus, these channels are pharmacological targets to treat neuropathic pain [65]. It has been proposed that ectopic discharges are associated with the development of spontaneous pain and mechanical allodynia [66]. Such spontaneous activities were generated in axons and soma of injured DRG neurons. There is evidence supporting the role of the hyperpolarization-activated current in ectopic discharges. The ectopic discharges are primarily generated from large- and medium-diameter DRG neurons in chronic neuropathic pain rats of the SNL, the chronic constriction injury for the sciatic nerve and the chronic compression for the DRG [67-69]. These findings strongly suggest that the hyperpolarization-activated currents are produced predominantly in these large-diameter and medium-diameter cells. It is generally known that activation time constants for HCN channels (ten to hundreds of milliseconds near the resting potential) are slower compared with the rapid activation rate for ectopic discharges (about 100 Hz) [70]. Thus, it is unlikely that the hyperpolarization-activated current participates in generation of ectopic discharges. However, in the dissociated DRG cells from rats subjected to chronic compression of the DRG, the time constant of fast activation of the hyperpolarization-activated current was significantly upregulated [71]. Therefore, it is conceivable that the hyperpolarization-activated currents are associated with the rapid ectopic discharges.
Conclusion
It has been evident that many ion channels contribute to generation of spontaneous pain. In addition, treatments for spontaneous pain targeting ion channels have grown rapidly. However, the molecular mechanisms by which drugs targeting ion channels relieve spontaneous pain remain largely unknown. Understanding these mechanisms will help to develop new therapeutic strategies for spontaneous pain.
Acknowledgement
This study was supported by JSPS KAKENHI Grant Number 17K08538.
Competing Interests
The author declares that he has no competing interests.
Article Info
Article Type
Review ArticlePublication history
Received: Mon 06, Aug 2018Accepted: Wed 22, Aug 2018
Published: Wed 05, Sep 2018
Copyright
© 2023 Hiroki Toyoda. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.NNB.2018.02.001
Author Info
Corresponding Author
Hiroki ToyodaDepartment of Oral Physiology, Osaka University Graduate School of Dentistry, Suita 565-0871, Japan
Figures & Tables
References
1. Baron R. (2006) Mechanisms of disease: neuropathic pain--a clinical perspective. Nat Clin Pract Neurol 2: 95-106. [Crossref]
2. Kleggetveit IP, Namer B, Schmidt R, Helas T, Ruckel M, et al. (2012) High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain 153: 2040-2047. [Crossref]
3. Lai J, Hunter JC, Porreca F. (2003) The role of voltage-gated sodium channels in neuropathic pain. Curr Opin Neurobiol 13: 291-297. [Crossref]
4. Catterall WA. (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13-25. [Crossref]
5. Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, et al. (2000) Nomenclature of voltage-gated sodium channels. Neuron 28: 365-368. [Crossref]
6. Novakovic SD, Eglen RM, Hunter JC. (2001) Regulation of Na+ channel distribution in the nervous system. Trends Neurosci 24: 473-478. [Crossref]
7. Devor M, Govrin-Lippmann R, Angelides K. (1993) Na+ channel immunolocalization in peripheral mammalian axons and changes following nerve injury and neuroma formation. J Neurosci 13: 1976-1992. [Crossref]
8. Matzner O, Devor M. (1994) Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J Neurophysiol 72: 349-359. [Crossref]
9. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. (2010) Sodium channels in normal and pathological pain. Annu Rev Neurosci 33: 325-347. [Crossref]
10. Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, et al. (2007) A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet 16: 2114-2121. [Crossref]
11. Estacion M, Han C, Choi JS, Hoeijmakers JG, Lauria G, et al. (2011) Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain 7: 92. [Crossref]
12. Yang Y, Wang Y, Li S, Xu Z, Li H, et al. (2004) Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 41: 171-174. [Crossref]
13. Renganathan M, Cummins TR, Waxman SG. (2001) Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol 86: 629-640. [Crossref]
14. Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, et al. (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321: 702-705. [Crossref]
15. Faber CG, Lauria G, Merkies IS, Cheng X, Han C, et al. (2012) Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A 109: 19444-19449. [Crossref]
16. Dib-Hajj S, Black JA, Cummins TR, Waxman SG. (2002) NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci 25: 253-259. [Crossref]
17. Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, et al. (2005) Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A 102: 9382-9387. [Crossref]
18. Lolignier S, Amsalem M, Maingret F, Padilla F, Gabriac M, et al. (2011) Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS One 6: e23083. [Crossref]
19. Deuis JR, Zimmermann K, Romanovsky AA, Possani LD, Cabot PJ, et al. (2013) An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1.6 in peripheral pain pathways. Pain 154: 1749-1757. [Crossref]
20. Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, et al. (2003) KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 23: 7227-7236. [Crossref]
21. Bayliss DA, Sirois JE, Talley EM. (2003) The TASK family: two-pore domain background K+ channels. Mol. Interv. 3(4): 205-219. [Crossref]
22. Armstrong CM. (2003) Voltage-gated K+ channels. Sci STKE 2003: re10. [Crossref]
23. Blank T, Nijholt I, Kye MJ, Spiess J. (2004) Small conductance Ca2+-activated K+ channels as targets of CNS drug development. Curr Drug Targets CNS Neurol Disord 3: 161-167. [Crossref]
24. Isomoto S, Kondo C, Kurachi Y. (1997) Inwardly rectifying potassium channels: their molecular heterogeneity and function. Jpn J Physiol 47: 11-39. [Crossref]
25. Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N. (2001) Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2: 175-184. [Crossref]
26. Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, et al. (2005) International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 57: 473-508. [Crossref]
27. Tsantoulas C, McMahon SB. (2014) Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci 37: 146-158. [Crossref]
28. Kim DS, Choi JO, Rim HD, Cho HJ. (2002) Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res 105: 146-152. [Crossref]
29. Tsantoulas C, Zhu L, Shaifta Y, Grist J, Ward JP, et al. (2012) Sensory neuron downregulation of the Kv9.1 potassium channel subunit mediates neuropathic pain following nerve injury. J Neurosci 32: 17502-17513. [Crossref]
30. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, et al. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291-366. [Crossref]
31. Butt AM, Kalsi A. (2006) Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med 10: 33-44. [Crossref]
32. Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. (2007) Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27: 11354-11365. [Crossref]
33. Tang X, Schmidt TM, Perez-Leighton CE, Kofuji P. (2010) Inwardly rectifying potassium channel Kir4.1 is responsible for the native inward potassium conductance of satellite glial cells in sensory ganglia. Neuroscience 166: 397-407. [Crossref]
34. Vit JP, Ohara PT, Bhargava A, Kelley K, Jasmin L. (2008) Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J Neurosci 28: 4161-4171. [Crossref]
35. Enyedi P, Czirjak G. (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559-605. [Crossref]
36. Acosta C, Djouhri L, Watkins R, Berry C, Bromage K, et al. (2014) TREK2 expressed selectively in IB4-binding C-fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci 34: 1494-1509. [Crossref]
37. Catterall WA Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3: a003947. [Crossref]
38. Chen E, Pare JF, Wichmann T, Smith Y (2017) Sub-synaptic localization of Cav3.1 T-type calcium channels in the thalamus of normal and parkinsonian monkeys. Brain Struct Funct 222: 735-748. [Crossref]
39. Kerr LM, Filloux F, Olivera BM, Jackson H, Wamsley JK (1988) Autoradiographic localization of calcium channels with [125I]omega-conotoxin in rat brain. Eur J Pharmacol 146: 181-183. [Crossref]
40. Mintz IM, Adams ME, Bean BP (1992) P-type calcium channels in rat central and peripheral neurons. Neuron 9: 85-95. [Crossref]
41. Miljanich GP, Ramachandran J (1995) Antagonists of neuronal calcium channels: structure, function, and therapeutic implications. Annu Rev Pharmacol Toxicol 35: 707-734. [Crossref]
42. Malmberg AB, Yaksh TL (1994) Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N- and P-type channels inhibits formalin-induced nociception. J Neurosci 14: 4882-4890. [Crossref]
43. Sluka KA (1998) Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammation. J Pharmacol Exp Ther 287: 232-237. [Crossref]
44. Matthews EA, Dickenson AH (2001) Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain 92: 235-246. [Crossref]
45. Rosenbaum T, Simon SA (2007) TRPV1 Receptors and Signal Transduction. [Crossref]
46. Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, et al. (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21: 531-543. [Crossref]
47. Zhou HY, Chen SR, Chen H, Pan HL. (2009) The glutamatergic nature of TRPV1-expressing neurons in the spinal dorsal horn. J Neurochem 108: 305-318. [Crossref]
48. Maione S, Starowicz K, Cristino L, Guida F, Palazzo E, et al. (2009) Functional interaction between TRPV1 and mu-opioid receptors in the descending antinociceptive pathway activates glutamate transmission and induces analgesia. J Neurophysiol 101: 2411-2422. [Crossref]
49. Cortright DN, Szallasi A. (2009) TRP channels and pain. Curr Pharm Des 15: 1736-1749. [Crossref]
50. Honore P, Chandran P, Hernandez G, Gauvin DM, Mikusa JP, et al. (2009) Repeated dosing of ABT-102, a potent and selective TRPV1 antagonist, enhances TRPV1-mediated analgesic activity in rodents, but attenuates antagonist-induced hyperthermia. Pain 142: 27-35. [Crossref]
51. Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, et al. (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112: 819-829. [Crossref]
52. Kwan KY, Glazer JM, Corey DP, Rice FL, Stucky CL (2009) TRPA1 modulates mechanotransduction in cutaneous sensory neurons. J Neurosci 29: 4808-4819. [Crossref]
53. Liang J, Bi H, Ji W (2010) Involvement of TRPA1 in ET-1-induced pain-like behavior in mice. Neuroreport 21: 201-205. [Crossref]
54. Wei H, Karimaa M, Korjamo T, Koivisto A, Pertovaara A (2012) Transient receptor potential ankyrin 1 ion channel contributes to guarding pain and mechanical hypersensitivity in a rat model of postoperative pain. Anesthesiology 117: 137-148. [Crossref]
55. Wemmie JA, Price MP, Welsh MJ (2006) Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci 29: 578-586. [Crossref]
56. Wemmie JA, Taugher RJ, Kreple CJ (2013) Acid-sensing ion channels in pain and disease. Nat Rev Neurosci 14: 461-471. [Crossref]
57. Hesselager M, Timmermann DB, Ahring PK (2004) pH Dependency and desensitization kinetics of heterologously expressed combinations of acid-sensing ion channel subunits. J Biol Chem 279: 11006-11015. [Crossref]
58. Frey Law LA, Sluka KA, McMullen T, Lee J, Arendt-Nielsen L, et al. (2008) Acidic buffer induced muscle pain evokes referred pain and mechanical hyperalgesia in humans. Pain 140: 254-264. [Crossref]
59. Voilley N, de Weille J, Mamet J, Lazdunski M (2001) Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci 21: 8026-8033. [Crossref]
60. Deval E, Gasull X, Noel J, Salinas M, Baron A, et al. (2010) Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther 128: 549-558. [Crossref]
61. Deval E, Baron A, Lingueglia E, Mazarguil H, Zajac JM, et al. (2003) Effects of neuropeptide SF and related peptides on acid sensing ion channel 3 and sensory neuron excitability. Neuropharmacology 44: 662-671. [Crossref]
62. Alvarez de la Rosa D, Zhang P, Shao D, White F, Canessa CM (2002) Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci U S A 99: 2326-2331. [Crossref]
63. Omori M, Yokoyama M, Matsuoka Y, Kobayashi H, Mizobuchi S, et al. (2008) Effects of selective spinal nerve ligation on acetic acid-induced nociceptive responses and ASIC3 immunoreactivity in the rat dorsal root ganglion. Brain Res 1219: 26-31. [Crossref]
64. Kaupp UB, Seifert R (2001) Molecular diversity of pacemaker ion channels. Annu Rev Physiol 63: 235-257. [Crossref]
65. Brown SM, Dubin AE, Chaplan SR (2004) The role of pacemaker currents in neuropathic pain. Pain Pract 4: 182-193. [Crossref]
66. Devor M, Raber P (1983) Autotomy after nerve injury and its relation to spontaneous discharge originating in nerve-end neuromas. Behav Neural Biol 37: 276-283. [Crossref]
67. Liu CN, Wall PD, Ben-Dor E, Michaelis M, Amir R, et al. (2000) Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 85: 503-521. [Crossref]
68. Kajander KC, Bennett GJ (1992) Onset of a painful peripheral neuropathy in rat: a partial and differential deafferentation and spontaneous discharge in A beta and A delta primary afferent neurons. J Neurophysiol 68: 734-744. [Crossref]
69. Song XJ, Hu SJ, Greenquist KW, Zhang JM, LaMotte RH (1999) Mechanical and thermal hyperalgesia and ectopic neuronal discharge after chronic compression of dorsal root ganglia. J Neurophysiol 82: 3347-3358. [Crossref]
70. Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, et al. (2006) The role of sodium channels in chronic inflammatory and neuropathic pain. J Pain 7: 1-29. [Crossref]
71. Yao H, Donnelly DF, Ma C, LaMotte RH (2003) Upregulation of the hyperpolarization-activated cation current after chronic compression of the dorsal root ganglion. J Neurosci 23: 2069-2074