Mechanisms of Cardiac Dysfunction in Heart Failure due to Myocardial Infarction
A B S T R A C T
Acute myocardial infarction (MI) is associated with marked elevation of plasma vasoactive hormones, ventricular arrhythmias, scar formation in the ischemic portion of left ventricle (LV) and hypertrophy of the viable LV as well as the right ventricle (RV). Particularly, elevated levels of plasma catecholamines and angiotensin II activate their membrane receptors and stimulate different signal transduction systems for producing cardiac hypertrophy, augmenting the activities of subcellular organelles and increasing cardiac function. While marked arrhythmias due to acute MI produce 30 to 40% mortality, hypertrophic alterations in the viable LV as well as RV are compensatory for maintaining hemodynamic homeostasis due to loss of cardiomyocytes. On the other hand, prolonged elevation of plasma vasoactive hormones in chronic MI produce deleterious effects on the hypertrophied heart by promoting the formation of oxyradicals, inducing Ca2+- handling abnormalities in subcellular organelles, depressing cardiac gene expression, activating different proteases and resulting in the development of cardiac dysfunction. Thus, in view of the complexities of mechanisms for both acute and chronic effects of MI, there is a real challenge of developing new interventions for preventing the transition of cardiac hypertrophy to heart failure as well as progression of the MI-induced cardiovascular abnormalities.
Keywords
Myocardial infarction, ventricular arrhythmias, cardiac hypertrophy, heart failure, vasoactive hormones, oxidative stress, Ca2+- handling abnormalities
Introduction
Congestive heart failure is a major health hazard, which is associated with cardiac dysfunction and increased mortality [1-7]. Depressed cardiac output due to inability of the heart to contract and relax properly occurs in heart failure, and different symptoms such as edema, breathlessness and intolerance to exercise become evident. Heart failure is invariably preceded by cardiac hypertrophy, which is an adaptive process to maintain heart function at initial stages of pathological stimulus [8-10]. It is commonly held that more than 700,000, deaths occur due to heart failure per year, costing the American economy about $50 billion annually. Several cardiovascular etiologies such as hypertension, diabetes, atherosclerosis, metabolic syndrome, valvular defects, aging, obesity, infective cardiomyopathy and myocardial infarction (MI) are known to result in heart failure [4, 10, 11]. Since MI, as a consequence of blockade of the coronary arteries (ischemic heart disease), is most prevalent among several cardiovascular abnormalities leading to the development of heart failure (Figure 1), this article is focused on discussion of the pathophysiology of MI-induced heart failure. In view of the complex nature of MI-induced effects on the heart, some of the events related to both acute and chronic actions will be discussed to highlight the progression of cardiovascular abnormalities. It is also planned to deal with a few pathogenic mechanisms of MI-induced cardiac hypertrophy as well as its transition to heart failure.
Acute MI-induced cardiovascular events
It is now well known that immediately after the induction of MI (blockade of the coronary blood flow), a portion of the left ventricle becomes ischemic and starts losing its contractile function. This event is associated with a reduction in cardiac output, a fall in blood pressure, activation of both sympathetic nervous system and renin-angiotensin system as well as pituitary system and release of a massive amount of catecholamines, angiotensin II and vasopressin in the circulation [12-17]. Increased plasma levels of other vasoactive hormones such as serotonin (due to activation of platelets) and endothelin (due to alterations in endothelium) have also been observed in acute MI. While elevated levels of these hormones serve as a compensatory mechanism to maintain blood pressure and cardiac performance, there occurs a defect in electrical conduction system in the heart due to ischemic portion of the left ventricle (Figure 2). The resultant ventricular arrhythmias are the cause for 30 to 40% mortality depending upon the size of ischemic area. The ischemic portion of the left ventricle starts becoming necrotic and ends up in the formation of scar tissue, which is fully healed within 3 to 4 weeks after coronary occlusion. Thus, acute MI is considered to be associated with scar formation in the left ventricle, elevated plasma levels of some vasoactive hormones, marked ventricular arrhythmias and high mortality. The development of cardiac hypertrophy as well as increase in cardiac function are also the major compensatory changes due to acute MI (Figure 3). These alterations are considered to occur mainly due to the elevated levels of circulating hormones such as catecholamines and angiotensin II, stimulation of different protein kinases and activation of different signal transduction pathways in the unifarcted myocardium [4, 10, 18-22]. Various protein kinases such as protein kinases A, Ca2+/calmodulin dependent protein kinase, protein kinase C and mitogen activated protein kinase are stimulated due to acute MI and promote different signal transduction systems for increasing the activities of subcellular organelles. It is noteworthy that the contractile function of the hypertrophied heart is not only augmented by the increased formation of contractile units but is also determined by the increased Ca2+-handling activities of subcellular organelles such as sarcolemma (SL), sarcoplasmic reticulum (SR) and myofibrils (MF).
In fact, increased functions of both SL and SR without any change in the MF activity have been observed in hypertrophied right ventricle during early phase of MI [23-29]. The hypertrophied right ventricle has been shown to exhibit hyperfunction and is considered to play a compensatory role during the development of contractile depression in the left ventricle due to MI [30]. The magnitude of cardiac hypertrophy and ventricular arrhythmias and mortality are considered to depend on scar size due to acute MI [31-34]. Electrocardiographic changes including ST-segment elevation, abnormal Q waves, premature ventricular complex, QTc prolongation and ventricular fibrillation have been shown to occur upon occlusion of the coronary artery. Since pretreatment of animals with an angiotensin-converting enzyme inhibitor and a serotonin-receptor antagonist were found to attenuate the MI-induced arrhythmias as well as mortality, it appears that excessive levels of circulating angiotensin II and serotonin may contribute in developing these fatal events due to myocardial ischemia [31, 33]. On the other hand, β1-adrenoceptor blocking agents failed to attenuate the occurrence or intensity of arrhythmias due to acute MI and accordingly, it was suggested that these alterations were due to the development of oxidative stress rather than the consequence of high levels of circulating catecholamines per se [34-36]. In fact, the MI-induced arrhythmias, scar size and mortality were markedly reduced by pretreatment with an antioxidant [32]. It should also be mentioned that there occurs some inflammatory response at the margin of the necrotic myocardium whereas the hypertrophied myocardium shows capillary growth, interstitial cell proliferation and accumulation of collagenous material during early phase of MI [37-40].
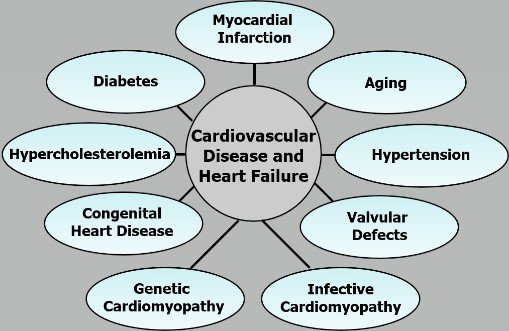
Figure 1: Schematic diagram of various etiologies of cardiovascular diseases and heart failure including myocardial infarction leading to the development of cardiovascular defects and heart failure.
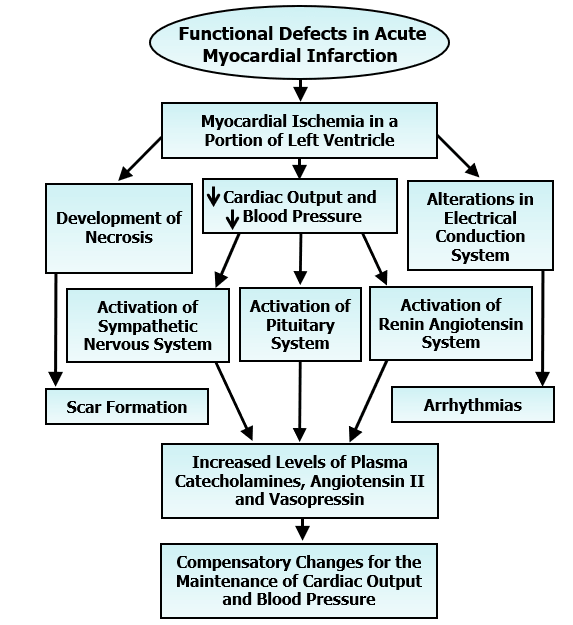
Figure 2: Sequence of events for the scar formation, occurrence of arrhythmias and development of compensatory changes in the heart subsequent to the induction of myocardial infarction.
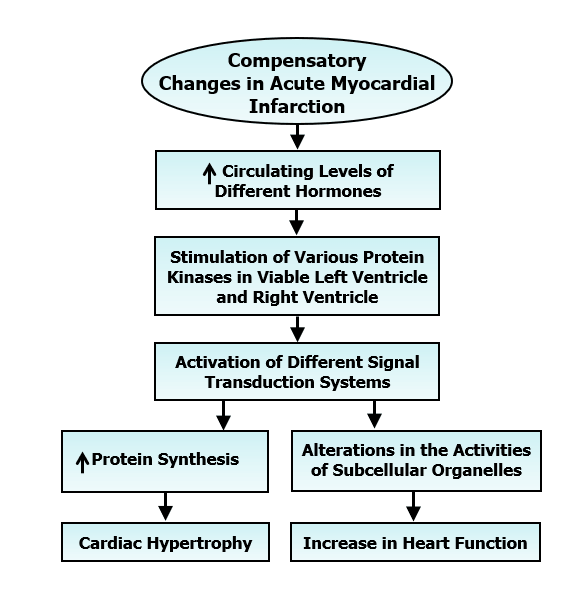
Figure 3: Signal transduction mechanisms for the development of cardiac hypertrophy and improvement of cardiac function subsequent to acute myocardial infarction.
Chronic MI-induced cardiovascular events
Although the acute MI is associated with some depression in the left ventricular + dP/dt and -dP/dt as well as some elevation in the left ventricular end-diastolic pressure, the overall cardiac performance is essentially unaltered due to the compensatory mechanisms such as cardiac hypertrophy of the viable left ventricle and the right ventricle [32, 33, 41]. On the other hand, there occurs a progressive depression of the left ventricular function associated with additional cardiac hypertrophy upon healing of the scar in 3 to 4 weeks after the induction of MI [4, 11, 42, 43]. These progression of changes in heart failure during the chronic phase of MI are accompanied by a progressive decline in both +dP/dt and - dP/dt as well as an increase in the left ventricular end diastolic pressure. All these alterations have been suggested to be a consequence of prolonged exposure of the heart to excessive levels of circulating vasoactive hormones such as catecholamines, angiotensin II, serotonin and endothelin (Figure 4). It should be pointed out that the role of catecholamines, angiotensin II, serotonin, endothelin and vasopressin in cardiovascular abnormalities due to chronic MI has been indicated elsewhere [4, 15, 44-47]. These vasoactive hormones activate their respective membrane receptors to increase the ventricular wall tension and stimulate signal transduction systems for producing additional cardiac hypertrophy. High levels of circulating catecholamines, serotonin and angiotensin II for a prolonged period upon oxidation are also known to generate oxyradicals to produce oxidative stress in the chronic phase of MI [4, 15, 44-48]. It should also be noted that alterations in endothelium not only release endothelin in the circulation but also increase the formation of NO, which combines with oxyradicals to produce nitrosative stress [4, 11, 43, 49]. Thus, both oxidative stress and nitrosative stress have been suggested to be intimately involved in the transition from cardiac hypertrophy to heart failure and cardiac dysfunction (Figure 4).
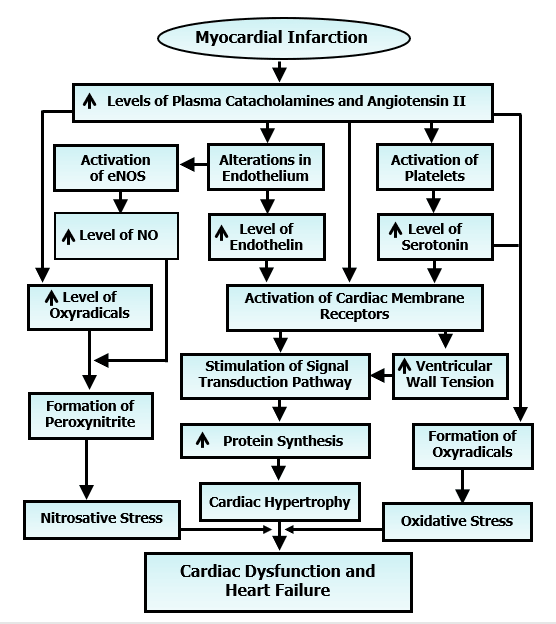
Figure 4: Role of some vasoactive hormones in the activation of membrane receptors for including cardiac hypertrophy as well as development of oxidative stress and nitrosative stress, which results in the transition of hypertrophy to heart failure due to myocardial infarction.
A wide variety of other cardiovascular events have also been considered to explain the occurrence of cardiac dysfunction in heart failure due to chronic MI [4 ,10, 11, 43]. There occurs inadequate growth of capillaries, which support oxygenation of the myocardium, and can be seen to induce functional hypoxia and cardiac dysfunction in chronic MI [38, 4]. Furthermore, disproportionate proliferation of non-myocyte cells and accumulation of collagenous proteins have been implicated in the development of MI-induced heart failure [4, 40, 50-52]. Elevated levels of pro-inflammatory cytokines and development of apoptosis have also been suggested to play an important role in MI-induced cardiac dysfunction. In addition, both cardiac remodeling and subcellular defects in Ca2+-handling are considered to be intimately associated with the progression of MI-induced heart failure [11, 42, 43, 49, 52-59].
MI-induced cardiac remodeling and subcellular defects
Some review articles on the cardiac remodeling and subcellular defects in MI-induced heart failure have appeared in the literature and it is generally believed that the term cardiac remodeling refers to changes in the shape and size of the heart [4, 11, 43]. Furthermore, subcellular defects concerning the organelles such as SL, SR, MF and mitochondria, which are intimately associated with cardiac contraction and relaxation cycle [4, 11, 42, 43, 50]. It was found that the ventricular wall becomes thickened for decreasing the wall tension and the cardiac output, ejection fraction and fractional shortening become reduced after the induction of MI. These MI-animals showed depressions in both left ventricular systolic and diastolic volumes as well as diameters indicating cardiac remodeling [60]. Hemodynamic assessment of MI-animals also revealed progressive decrease in left ventricular pressure as well as both systolic and diastolic blood pressure. These MI-induced changes in cardiac remodeling and cardiac function were attenuated by angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, β-adrenoceptors blocking agents, 5-HT receptor antagonists and metabolic inhibitors suggesting that angiotensin II, catecholamines, serotonin and metabolic derangements are involved in the MI-induced cardiac remodeling and heart dysfunction [22, 27, 28, 45, 46, 61-69]. It has also been demonstrated that SL genes, proteins and Ca2+-transporting activities are altered during the development of MI-induced heart failure [27, 28, 43, 61, 69-71]. The SL defects in MI-failing hearts were prevented by metabolic inhibitors, β-adrenoceptors blockade and renin-angiotensin blockade [43, 52, 61, 63, 69]. Likewise, the MI-induced alterations in myosin gene expression, protein content and MF Ca2+-stimulated ATPase activities were attenuated by blockade of the renin angiotensin system, serotonin-antagonists and β-adrenoceptors blockade [29, 43, 64, 68, 72]. Furthermore MI-induced changes in SR gene expression, protein content, Ca2+-pump and Ca2+-release activities were prevented by treatment with renin-angiotensin blockers, β-adrenoceptors blockers and serotonin antagonists [28, 32, 40, 43, 62, 65, 68, 69, 73]. The beneficial effects of catecholamine-receptor antagonists, angiotensin-receptor blockers, serotonin antagonists, vasopressin blockers, endothelin receptor blockers and cytokines on MI-induced changes in heart function, cardiac remodeling subcellular activities have been reviewed earlier [16, 43, 45, 46, 49, 53].
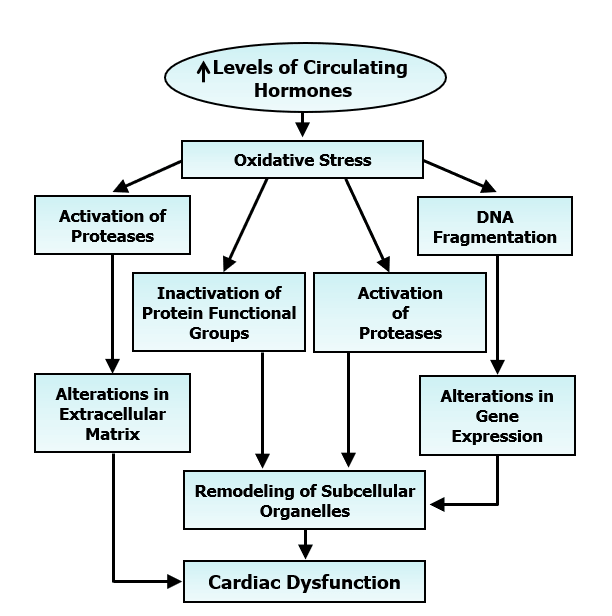
Figure 5: Role of prolonged increase in the level of some vasoactive hormones in the development of oxidative stress for inducing cardiac dysfunction due to changes in extracellular matrix, gene expression and remodelling of subcellular organelles.
Role of oxidative stress and Ca2+-handling abnormalities in MI-induced cardiac dysfunction
The adverse chronic effects of MI are related to progressive development of heart failure as a consequence of prolonged exposure of the heart to elevated levels of several vasoactive hormones including catecholamines and angiotensin II in the circulation [4, 11, 44]. Although heart failure patients are more prone to sudden cardiac death and high mortality, this article is not intended to discuss this topic [36, 73]. There is evidence to suggest an increased formation of oxyradicals and reactive oxygen species leading to the development of oxidative stress upon the oxidation of catecholamines and serotonin whereas angiotensin II promotes the development of oxidative stress by activating the NADPH oxidase for the production of oxyradicals [36, 48, 74-76]. Furthermore, elevated levels of circulating vasoactive hormones are known to promote the entry of Ca2+ into cardiomyocytes and cause the occurrence of intracellular Ca2+-overload [36, 77, 78]. Accordingly, it is planned to deal with the pathophysiology of MI-induced cardiac dysfunction involving the development of oxidative stress and occurrence of intracellular Ca2+-overload in the genesis of subcellular defects. It is becoming evident that oxidative stress is not only a major mechanism for the transition of MI-induced cardiac hypertrophy to heart failure but is also intimately involved in the progression of cardiac remodeling and heart failure [74]. It is generated by the action elevated levels of different hormones as well as due to functional hypoxia in the hypertrophied viable myocardium. An increase in pro-inflammatory cytokines has also been suggested to generate oxidative stress [53]. The development of oxidative stress can be seen to induce cardiac dysfunction by increasing the activities of various proteases and subsequent alterations in the extracellular matrix and the function of subcellular organelles [4, 42, 43, 52, 79]. It is noteworthy that oxidative stress can alter the activities of the different subcellular organelles by the depressing cardiac gene expression due DNA fragmentation as well as inactivation of the functional groups of the subcellular proteins. A schematic diagram showing all these events for the oxidative stress-induced cardiac dysfunction is given in (Figure 5). Increased development of oxidative stress has also been observed in patients with congestive heart failure [80-83].
Another mechanism for the development of MI-induced cardiac dysfunction and heart failure is the occurrence of Ca2+-handling abnormalities in the viable left ventricle and increased concentration of intracellular Ca2+ [4, 42, 43, 52]. The development of increase in intracellular [Ca2+]𝒊 due to increased entry of Ca2+ in cardiomyocyte by the activation of membrane receptors for vasoactive hormones as well as depressed Na+- Ca2+ exchange and Na+- K+ ATPase, can be seen to produce mitochondrial Ca2+-overload, depress ATP production and deplete of energy stores in the myocardium [4, 11]. The depletion of energy stores can also be produced by increased ATP hydrolysis. Furthermore, intracellular Ca2+-overload has been shown to produce subcellular remodeling by increasing the activities of different proteases as well as by depressing the cardiac gene expression [79, 84]. Thus, both depletion of energy stores and subcellular remodeling have been suggested to explain cardiac dysfunction due to the occurrence of intracellular Ca2+-overload. These events have been depicted in (Figure 6). It should also be pointed out the development of intracellular Ca2+-overload has been shown to induce apoptosis in cardiomyocytes, which may be associated with the development of cardiac dysfunction and heart failure [85]. Due to the interactive nature of intracellular Ca2+-overload and oxidative stress, it is difficult to determine the individual contribution of these pathogenic factors for the chronic MI-induced heart failure

Figure 6: Role of prolonged increase in the level of some vasoactive hormones in the development of increase in intracellular Ca2+ for inducing cardiac dysfunction due to changes in gene expression, increase in proteolysis, decrease in ATP production and depletion of energy stores.
Conclusion
A complex set of events is considered to occur during the development of MI-induced cardiovascular abnormalities. During the acute stage of MI, loss of cardiomyocytes in the ischemic portion of the left ventricle results in scar formation, massive release of several vasoactive hormones, marked arrhythmias, increased mortality and development of hypertrophy of the non-ischemic viable myocardium. While the deleterious effects of acute MI are associated with structural abnormalities due to myocardial ischemia, the compensatory effects at early stage of MI are elicited mainly through the activation of membrane receptors for different hormones as well as stimulation of various signal transduction pathways. On the other hand, the chronic phase of MI (which starts after the healing of scar) is associated with development of heart failure as a consequence of prolonged exposure to high levels of circulating vasoactive hormones. The transition of cardiac hypertrophy to heart failure seem to be a consequence of functional hypoxia due to the loss of capillary growth, disproportionate proliferation of non-myocytes, accumulation of collagenous proteins, development of cardiac apoptosis and formation of proinflammatory cytokines. In addition, cardiac remodeling, activation of different proteolytic enzymes, subcellular defects for Ca2+-handling, and occurrence of oxidative stress as well as increased concentration of intracellular Ca2+ have also been implicated in the progression of MI-induced cardiac dysfunction and heart failure. Although most of the existing therapies for MI-induced cardiovascular abnormalities are based on either lowering the elevated levels of circulating vasoactive hormones or their receptor antagonism, there is a great challenge for developing newer strategies to further improve the treatment of heart failure.
Acknowledgements
The infrastructure support for this project was provided the St. Boniface Hospital Research Foundation, Winnipeg, Canada.
Conflict of interest
The authors declare that there was no conflict of interest.
Article Info
Article Type
Research ArticlePublication history
Received: Sat 23, Nov 2019Accepted: Fri 06, Dec 2019
Published: Mon 16, Dec 2019
Copyright
© 2023 Naranjan S. Dhalla. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JICOA.2019.04.12
Figures & Tables
References
- Brophy JM (1992) Epidemiology of congestive heart failure: Canadian data from 1970 to 1989. Can J Cardiol 8: 495-498. [Crossref]
- Smith WM (1985) Epidemiology of congestive heart failure. Am J Cardiol 55: 3A-8A. [Crossref]
- Parmley WW (1985) Pathophysiology of congestive heart failure. Am J Cardiol 56: 7A-11A. [Crossref]
- Dhalla NS, Afzal N, Beamish RE, Naimark B, Takeda N et al. (1993) Pathophysiology of cardiac dysfunction in congestive heart failure. Can J Cardiol 9: 873-887. [Crossref]
- Cohn JN, Ferrari R, Sharpe N (2000) Cardiac remodeling-concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35: 569-582. [Crossref]
- Mudd JO, Kass DA (2008) Tackling heart failure in the twenty-first century. Nature 451: 919- 928. [Crossref]
- Jessup M, Brozena S (2003) Heart failure. N Engl J Med 348: 2007-2018. [Crossref]
- Grossman W (1980) Cardiac hypertrophy: useful adaptation or pathologic process? Am J Med 69: 576-584. [Crossref]
- Wikman Coffelt J, Parmley WW, Mason DT (1979) The cardiac hypertrophy process. Analyses of factors determining pathological vs. physiological development. Circ Res 45: 697-707. [Crossref]
- Dhalla NS, Dent MR, Tappia PS, Sethi R, Barta J et al. (2006) Subcellular remodeling as a viable target for the treatment of congestive heart failure. J Cardiovasc Pharmacol Ther 11: 31-45. [Crossref]
- Dhalla NS, Rangi S, Babick AP, Zieroth S, Elimban V (2012) Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail Rev 17: 671-681. [Crossref]
- Francis GS (1986) Development of arrhythmias in the patient with congestive heart failure: pathophysiology, prevalence and prognosis. Am J Cardiol 57: 3B-7B. [Crossref]
- Packer M (1988) Neurohormonal interactions and adaptations in congestive heart failure. Circulation 77: 721-730. [Crossref]
- Lee CS, Tkacs NC (2008) Current concepts of neurohormonal activation in heart failure: mediators and mechanisms. AACN Adv Crit Care 19: 364-385. [Crossref]
- Zhang W, Elimban V, Nijjar MS, Gupta SK, Dhalla NS (2005) Role of renin-angiotensin system in the development of cardiac hypertrophy and heart failure. In: Adaptation Biology and Medicine: Current Concepts, Volume 4, Ed. by A Hargens, N Takeda and PK Singal, Narosa Publishing House, New Delhi, 239-257.
- Rehsia NS, Dhalla NS (2010) Mechanisms of the beneficial effects of β-adrenoceptor antagonists in congestive heart failure. Exp Clin Cardiol 15: e86-e95. [Crossref]
- Rehsia NS, Dhalla NS (2010) Potential of endothelin-1 and vasopressin antagonists for the treatment of congestive heart failure. Heart Fail Rev 15: 85-101. [Crossref]
- Wang J, Liu X, Arneja AS, Dhalla NS (1999) Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can J Cardiol 15: 683-690. [Crossref]
- English JM, Cobb MH (2002) Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci 23: 40-45. [Crossref]
- Zhang W, Elimban V, Nijjar MS, Gupta SK, Dhalla NS (2003) Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp Clin Cardiol 8: 173-183. [Crossref]
- Kirchhefer U, Schmitz W, Scholz H, Neumann J (1999) Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res 42: 254-261. [Crossref]
- Netticadan T, Temsah RM, Kawabata K, Dhalla NS (2000) Sarcoplasmic reticulum Ca2+/Calmodulin-dependent protein kinase is altered in heart failure. Circ Res 86: 596-605. [Crossref]
- Sethi R, Dhalla KS, Beamish RE, Dhalla NS (1997) Differential changes in left and right ventricular adenylyl cyclase activities in congestive heart failure. Am J Physiol 272: H884 -H893. [Crossref]
- Ganguly PK, Dhalla KS, Shao Q, Beamish RE, Dhalla NS (1997) Differential changes in sympathetic activity in left and right ventricles in congestive heart failure following myocardial infarction. Am Heart J 133: 340-345. [Crossref]
- Sethi R, Elimban V, Chapman D, Dixon IMC, Dhalla NS (1998) Differential alterations in left and right ventricular G-proteins in congestive heart failure due to myocardial infarction. J Mol Cell Cardiol 30: 2153-2163. [Crossref]
- Sethi R, Saini HK, Wang X, Elimban V, Babick A et al. (2006) Differential changes in β-adrenoceptor signal transduction in left and right ventricles of infarcted rats. Can J Physiol Pharmacol 84: 747-754. [Crossref]
- Dixon IMC, Hata T, Dhalla NS (1992) Sarcolemmal Na+-K+-ATPase activity in congestive heart failure due to myocardial infarction. Am J Physiol 262: C664-C671. [Crossref]
- Afzal N, Dhalla NS (1992) Differential changes in left and right ventricular SR calcium transport in congestive heart failure. Am J Physiol 262: H868-H874. [Crossref]
- Wang J, Liu X, Ren B, Rupp H, Takeda N (2002) Modification of myosin gene expression by imidapril in failing heart due to myocardial infarction. J Mol Cell Cardiol 34: 847-857. [Crossref]
- Zimmer HG, Gerdes AM, Lortel S, Mell G (1990) Changes in heart function and cardiac cell size in rats with chronic myocardial infarction. J Mol Cell Cardiol 22: 1231-1243. [Crossref]
- Ren B, Lukas A, Shao Q, Guo M, Takeda N et al. (1998) Electrocardiographic changes and mortality due to myocardial infarction in rats with or without imidapril treatment. J Cardiovasc Pharmacol Ther 3: 11-22. [Crossref]
- Sethi R, Takeda N, Nagano M, Dhalla NS (2000) Beneficial effects of vitamin E treatment in acute myocardial infarction. J Cardiovasc Pharmacol Ther 5: 51-58. [Crossref]
- Brasil D, Temsah RM, Kumar K, Kumamoto H, Takeda N et al. (2002) Blockade of 5 HT2A receptors by sarpogrelate protects the heart against myocardial infarction in rat. J Cardiovasc Pharmacol Ther 7: 53-59. [Crossref]
- Adameova AD, Bhullar SK, Elimban V, Dhalla NS (2018) Activation of β1-adrenoceptors may not be involved in arrhythmogenesis in ischemic heart disease. Rev Cardiovasc Med 19: 97-101. [Crossref]
- Daugherty A, Frayn KN, Redfern WS, Woodward B (1986) The role of catecholamines in the production of ischaemia-induced ventricular arrhythmias in the rat in vivo and in vitro. Br J Pharmacol 87: 265-277. [Crossref]
- Dhalla NS, Adameova A, Kaur M (2010) Role of catecholamine oxidation in sudden cardiac death. Fund Clin Pharmacol 24: 539-546. [Crossref]
- Fishbein MC, Maclean D, Maroko PR (1978) Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution. Am J Pathol 90: 57-70. [Crossref]
- Anversa P, Beghi C, Kikkawa Y, Olivetti G (1986) Myocardial infarction in rats. Infarct size, myocyte hypertrophy, and capillary growth. Circ Res 58: 26-37. [Crossref]
- Anversa P, Beghi C, McDonald SL, Levicky V, Kikkawa Y et al. (1984) Morphometry of right ventricular hypertrophy induced by myocardial infarction in the rat. Am J Pathol 116: 504-513. [Crossref]
- Pelouch V, Dixon IMC, Sethi R, Dhalla NS (1993) Alteration of collagenous protein profile in congestive heart failure secondary to myocardial infarction. Mol Cell Biochem 129: 121‑131. [Crossref]
- Shao Q, Takeda N, Temsah R, Dhalla KS, Dhalla NS (1996) Prevention of hemodynamic changes due to myocardial infarction by early treatment of rats with imidapril. Cardiovasc Pathobiol 1: 180-186.
- Dhalla NS, Shao Q, Panagia V (1998) Remodeling of cardiac membranes during the development of congestive heart failure. Heart Fail Rev 2: 261-272.
- Dhalla NS, Dent MR, Tappia PS, Sethi R, Barta J et al. (2006) Subcellular remodeling as a viable target for the treatment of congestive heart failure. J Cardiovasc Pharmacol Ther 11: 31-45. [Crossref]
- Vikenes K, Farstad M, Nordrehaug JE (1999) Serotonin is associated with coronary artery disease and cardiac events. Circulation 100: 483-489. [Crossref]
- Guo X, Saini HK, Wang J, Gupta SK, Goyal RK et al. (2005) Prevention of remodeling in congestive heart failure due to myocardial infarction by blockade of renin angiotensin system. Expert Rev Cardiovasc Ther 3: 717-732. [Crossref]
- Machackova J, Sanganalmath SK, Barta J, Dhalla KS, Dhalla NS (2010) Amelioration of cardiac remodeling in congestive heart failure by β-adrenoceptor blockade is associated with depression in sympathetic activity. Cardiovasc Toxicol 10: 9-16. [Crossref]
- Adameova A, Abdellatif Y, Dhalla NS (2009) Role of the excessive amounts of circulating catecholamines and glucocorticoids in stress-induced heart disease. Can J Physiol Pharmacol 87: 493-514. [Crossref]
- Mialet Perez J, Santin Y, Parini A (2018) Monoamine oxidase-A, serotonin and norepinephrine: synergistic players in cardiac physiology and pathology. J Neural Transm (Vienna) 125: 1627-1634. [Crossref]
- Xu B, Li H (2015) Brain mechanisms of sympathetic activation in heart failure: Roles of the renin‑angiotensin system, nitric oxide and pro‑inflammatory cytokines (Review). Mol Med Rep 12: 7823-7829. [Crossref]
- Weber KT, Brilla CG (1991) Pathological hypertrophy and cardiac interstitium-fibrosis and renin-angiotensin-aldosterone system. Circulation 83: 1849-1865. [Crossref]
- Ju H, Zhaoe S, Jassal DS, Dixon IM (1997) Effect of AT1 receptor blockade on cardiac collagen remodeling after myocardial infarction. Cardiovasc Res 35: 223-232. [Crossref]
- Dhalla NS, Saini Chohan HK, Rodriguez Leyva D, Elimban V, Dent MR et al. (2009) Subcellular remodeling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res 81: 429-438. [Crossref]
- Bartekova M, Radosinska J, Jelemensky M, Dhalla NS (2018) Role of cytokines and inflammation in heart function during health and disease. Heart Fail Rev 23: 733-758. [Crossref]
- Kaur K, Sharma AK, Singal PK (2006) Significance of changes in TNF-alpha and IL-10 levels in the progression of heart failure subsequent to myocardial infarction. Am J Physiol Heart Circ Physiol 291: H106-H113. [Crossref]
- Dhingra S, Sharma AK, Singla DK, Singal PK (2007) p38 and ERK1/2 MAPKs mediate the interplay of TNF-alpha and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol 293: H3524-H3531. [Crossref]
- Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK (2009) IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res 82: 59-66. [Crossref]
- Sharma SK, Chapman D, Temsah R, Netticadan T, Brasil DP et al. (1999) Prevention of Vascular Apoptosis in Myocardial Infarction by Losartan. J Cardiovasc Pharmacol Ther 4: 77-84. [Crossref]
- Li Z, Bing OH, Long X, Robinson KG, Lakatta EG (1997) Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol 272: H2313-H2319. [Crossref]
- MacLellan WR, Schneider MD (1997) Death by design. Programmed cell death in cardiovascular biology and disease. Circ Res 81: 137-144. [Crossref]
- Sanganalmath SK, Barta J, Takeda N, Kumamoto H, Dhalla NS (2008) Antiplatelet therapy mitigates cardiac remodeling and dysfunction in congestive heart failure due to myocardial infarction. Can J Physiol Pharmacol 86: 180-189. [Crossref]
- Sethi R, Dhalla KS, Ganguly PK, Ferrari R, Dhalla NS (1999) Beneficial effects of propionyl L carnitine on sarcolemmal changes in heart failure due to myocardial infarction. Cardiovasc Res 42: 607-615. [Crossref]
- Shao Q, Ren B, Zarain Herzberg A, Ganguly PK, Dhalla NS (1999) Captopril treatment improves the sarcoplasmic reticular Ca2+-transport in heart failure due to myocardial infarction. J Mol Cell Cardiol 31: 1663-1672. [Crossref]
- Sethi R, Wang X, Ferrari R and Dhalla NS (2004) Improvement of cardiac function and β-adrenergic signal transduction by propionyl L-carnitine in congestive heart failure due to myocardial infarction. Coronary Art Disease 15: 65-71. [Crossref]
- Wang J, Guo X, Dhalla NS (2004) Modification of myosin protein and gene expression in failing hearts due to myocardial infarction by enalapril or losartan. Biochim Biophys Acta 1690: 177-184. [Crossref]
- Shao Q, Ren B, Saini HK, Netticadan T, Takeda N et al. (2005) Sarcoplasmic reticulum Ca2+-transport and gene expression in congestive heart failure are modified by imidapril treatment. Am J Physiol Heart Circ Physiol 288: H1674-H1682. [Crossref]
- Shao Q, Ren B, Elimban V, Tappia PS, Takeda N et al. (2005) Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am J Physiol Heart Circ Physiol 288: H2637-H2646. [Crossref]
- Babick AP, Dhalla NS (2007) Role of subcellular remodeling in cardiac dysfunction due to congestive heart failure. Med Princ Pract 16: 81-89. [Crossref]
- Sanganalmath SK, Babick AP, Barta J, Kumamoto H, Takeda N et al. (2008) Antiplatelet therapy attenuates subcellular remodelling in congestive heart failure. J Cell Mol Med 12: 1728-1738. [Crossref]
- Guo X, Wang J, Elimban V, Dhalla NS (2008) Both enalapril and losartan attenuate sarcolemmal Na+-K+-ATPase remodeling in failing rat heart due to myocardial infarction. Can J Physiol Pharmacol 86: 139-147. [Crossref]
- Dixon IM, Lee SL, Dhalla NS (1990) Nitrendipine binding in congestive heart failure due to myocardial infarction. Circ Res 66: 782-788. [Crossref]
- Dixon IM, Hata T, Dhalla NS (1992) Sarcolemmal Ca2+-transport in congestive heart failure due to myocardial infarction in rats. Am J Physiol 262: H1387-H1394. [Crossref]
- Machackova J, Sanganalmath SK, Elimban V, Dhalla NS (2011) β-adrenergic blockade attenuates cardiac dysfunction and myofibrillar remodeling in congestive heart failure. J Cell Mol Med 15: 545-554. [Crossref]
- Dhalla NS (2018) Formation of aminochrome leads to cardiac dysfunction and sudden cardiac death. Circ Res 123: 409-411. [Crossref]
- Dhalla NS, Temsah RM, Netticadan T (2000) Role of oxidative stress in cardiovascular diseases. J Hypertension 18: 655-673. [Crossref]
- Li B, Tian J, Sun Y, Xu TR, Chi RF et al. (2015) Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim Biophys Acta 1852: 805-815. [Crossref]
- Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM (2013) Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal 19: 1110-1120. [Crossref]
- Shao Q, Saward L, Zahradka P, Dhalla NS (1998) Ca2+ mobilization in adult rat ardiomyocytes by angiotensin type I and 2 receptors. Biochem Pharmacol 55: 1413-1418. [Crossref]
- Golfman LS, Hata T, Beamish RE, Dhalla NS (1993) Role of endothelin in heart function in health and disease. Can J Cardiol 9: 635-653. [Crossref]
- Müller AL, Dhalla NS (2012) Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail Rev 17: 395-409. [Crossref]
- Chen L, Zang Y, Bai B, Zhu M, Zhao B et al. (1992) Electron spin resonance determination and superoxide dismutase activity in polymorpho-nuclear leukocytes in congestive heart failure. Can J Cardiol 8 :756 -760. [Crossref]
- Ghatak A, Brar MJ, Agarwal A, Goel N, Rastogi AK et al. (1996) Oxyfree radical system in heart failure and therapeutic role of oral vitamin E. Int J Cardiol 57: 119 -127. [Crossref]
- Diaz Velez CR, Garcia Castineiras S, Mendoza Ramos E, Hernandez Lopez E (1996) Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am Heart J 131: 146-152. [Crossref]
- Belch JJ, Bridges AB, Scott N, Chopra M (1991) Oxygen free radicals and congestive heart failure. Br Heart J 65: 245-248. [Crossref]
- Ozcelikay AT, Chapman D, Elimban V, Dhalla NS (2014) Role of intracellular Ca2+-overload in inducing changes in cardiac gene expression. Curr Res Cardiol 1: 13-16.
- Xu Y J, Saini HK, Zhang M, Elimban V, Dhalla NS (2006) MAPK activation and apoptotic alterations in hearts subjected to calcium paradox are attenuated by taurine. Cardiovasc Res 72: 163-174. [Crossref]