Neurogenesis and Oligodendrogenesis in a Mouse Model of Blast-Induced Traumatic Brain Injury
A B S T R A C T
Neurological manifestations of blast-induced Post Traumatic Stress Disorder (PTSD) extend long after the initial injury indicating lasting changes in brain function. In this study, we characterized brain injury, changes in neurogenesis and oligodendrogenesis in an adult murine blast model following a short (5 days) and long (21 days) post-blast recovery. Acoustic blasts led to an initial, activation of microglia and astrogliosis and a widespread cortical and subcortical apoptosis. The loss of myelinated cortical axons at 5 days was followed by the reappearance of abnormal misdirected fibers at 21 days. At 21 days post-blast, we observed increases in doublecortin-positive (DCX+) neuroblasts in the subventricular zone (SVZ) and hippocampal subgranular zone (SGZ) indicating increased neurogenesis. No changes in DCX+ cells were found in the brain cortex. In the cortex, the early disappearance of myelinated neuronal fibers was accompanied by a loss of O4+ oligodendrocytes and their Ki67-expreasing (Ki67+) oligodendrocyte precursor cells (OPC). However, at 5 days we observed a robust appearance of cells expressing Olig2 (O2+), an early determinant of oligodendrocyte lineage. At 21 days post-blast, the population of OPC increased and the mature O4+ oligodendrocytes were restored to control levels. In contrast, in the SVZ and SGZ, O4+ cells were not affected by the blast suggesting a local cortical origin for cortical oligodendrogenesis. These results suggest that blast-induced activation of SVZ and SGZ neurogenesis and cortical oligodendrogenesis could have long-lasting impact on brain function including memory disorders observed in both animal models and human’s PTSD.
Keywords
Blast induced traumatic brain injury, traumatic brain injury, neurogenesis, oligodendrogenesis
Introduction
As technology has evolved, so has that traumatic power of weapons used in warfare. While improvements in protective armor and post-trauma care have increased the survival of soldiers in the battlefield, new types of the injuries have emerged among survivors. The use of improvised explosive devices (IEDs) such as roadside bombs have dramatically increased in the past few decades [1, 2]. What makes the IEDs so dangerous is their unpredictability and the large number of soldiers and civilians affected. The trauma from IEDs is multifactorial, including shockwave-induced haemorrhage, damage to internal organs, injuries caused by projectiles, and rapid acceleration/deceleration of the body. Among the most serious and long-lasting injuries is blast-induced traumatic brain injury (bTBI) [3].
According to the Defense and Veteran Brain Injury Center, from 2000-2018 there have been 383,947 US service members diagnosed with TBI [4]. TBI is characterized by an acute loss of consciousness typically for more than 30 minutes, followed by a confused or disoriented state that lasts less than 24 hours, and/or an initial memory loss lasting less than 24 hours [5-7]. Because of the proliferation of IED, bTBI has been referred to as the signature injury of recent military conflicts in the Middle East [8]. A bTBI can be divided into four biomechanically distinct phases. The primary phase results from physical forces emanating from the explosive with rapid transmission of the pressure wave through brain tissue and its vasculature. The secondary phase typically results from projectiles penetrating the head. The tertiary phase results from head contact/acceleration forces when the body is displaced by the massive blast pressure wave. The quaternary phase incorporates haemorrhagic shock, and chemical or thermal burns [9-13]. The vast majority of bTBI occurs in the first three phases.
In a study conducted on postmortem brains subjected to blast waves, simulations showed significant shear and dilatational energies develop in the brain within 4-5 milliseconds after blast wave exposure. These changes were not randomly distributed, but developed in specific locations such as frontal lobes, upper brain stem, and internal capsule [14]. Both the direct shock wave and the indirect transfer of the shock wave through blood vessels damaged the brain tissue and caused neurological impairments [15, 16]. Blast waves initiate a cascade of cellular pathological processes, which damage the microvasculature and blood-brain barrier [17, 18]. Breakdown of the blood-brain barrier leads to brain edema, which increases intracranial pressure, impairs cerebral oxygenation and causes neuro-inflammation [19, 20].
Goldstein et al. (2012) performed post-mortem analysis of the brains of four military veterans who had a history of blast exposure. All brains showed tau pathology in several cortical regions accompanied by axon dystrophy and degeneration as well as clusters of activated microglia [21]. The same research team employed a mouse model of TBI in which they observed similar signs such as neuro-inflammation in cortex, hippocampus, cerebellum, brainstem and corticospinal tract, phosphorylated tau neuropathology in superficial cortical layers and hippocampus, neurodegeneration in the hippocampus, activated perivascular microglia throughout the brain, and loss of cerebellar Purkinje cells [21]. Arun et al. (2013) studied mitochondrial energy metabolism both in vivo in a mouse bTBI model and in a human neuroblastoma culture subjected to a single or three blast waves. Single or multiple blasts decreased neuronal ATP levels 6 hours post-blast, indicating a severe acute neuronal mitochondrial dysfunction. The decrease in ATP correlated with the reduced expression of GOT2-encoded mitochondrial aspartate aminotransferase, which in neurons plays an important role in metabolism and energy production [22]. These mitochondrial changes have also been observed in the rat hippocampus suggesting that that the neurodegenerative process was a consequence of oxidative stress and mitochondrial dysfunction [23].
In a mouse TBI study, blood-borne macrophages entered brain through the damaged blood-brain barrier accompanied by an activation of the endogenous microglia [24]. These immune cells accumulated at the site of injury where they phagocytosed cellular debris and foreign bodies and took part in the inflammation, promoting and directing tissue repair and maintaining homeostasis [24]. Activated microglia on the other hand release cytotoxic and pro-inflammatory cytokines, interferon gamma (IFNγ) or tumor necrosis factor (TNFα), which promotes the death of proliferating oligodendrocyte precursor cells and mature oligodendrocytes, respectively [25, 26]. Trauma-induced reactive astrocytes are known to impair the long-term recovery, repair of white matter and remyelination. Reactive astrocytes secrete TNFβ and interleukinβ that inhibit regrowth of axons thereby impeding the migration of OPC and promoting the death of oligodendrocytes [27, 28].
Currently, it is unknown whether bTBI disrupts neuronal or oligodendrocyte progenitor cells leading to impaired regeneration or maladaptive plasticity. In adult brain, stem/progenitor cells reside mainly within two niches, the subcortical sub-ventricular zone (SVZ) and hippocampal sub-granular zone (SGZ). The SVZ brain stem/progenitor cells can give rise to neuronal or glial cell types that migrate to the cortex while the SGZ repopulate the hippocampal dental gyrus.
The SVZ, which lies along the walls of the lateral ventricle, is the largest germinal zone in the adult mammalian brain [29]. The neural stem/progenitor cells in the SVZ form at least four different cell types defined by their morphology, ultrastructure, and molecular markers [30]. The cellular characteristics and locations of two of these cells, type-B and type-C cells, suggest that one or both of these could be involved in generating new neurons and oligodendrocytes. Type B cells become the primary precursors of new neurons, although, they are also capable of generating small numbers of nonmyelinating NG2+ OPCs and mature myelinating oligodendrocytes [30-33]. Type-A neuroblasts have been shown to migrate anteriorly from the SVZ via the rostral migratory stream toward the olfactory bulb (OB), where they differentiate into local interneurons [34-38]. These cells may also migrate to the cerebral cortex following brain injury and reactivate developmental neurogenic Integrative Nuclear FGFR1 Signaling mechanism [39]. A small fraction of type-C cells generates OPCs, which migrate radially out of the SVZ into the overlying white matter and cortex [32, 33, 40].
Neurogenesis in the hippocampal SGZ has been linked to the formation of memory. As many as 250,000 new neurons are incorporated into the rodent dentate gyrus every month or 6% of its total cell number [41]. The SGZ also has a heterogeneous population of stem/progenitor cells. Type 1 cells, multipotent largely quiescent stem cells, resides in the hippocampus [42]. Type 1 cells, also referred to as radial astrocytes, type-1 progenitors or radial glia-like cells, generate new dentate granule neurons via IPC1 and IPC2 (also known as type 2a and 2b cells respectively) [41-46].
Through asymmetric divisions, radial astrocytes replenish themselves and generate the amplifying neural progenitors (ANPs) that produce the D cells, the intermediate precursors of new granule neurons [42, 47]. These D cells express the polysialylated form of the neural cell adhesion molecule (PSA-NCAM) as well as DCX associated with the neuroblast phenotype that migrate and differentiate into neurons [41, 48, 49].
Exposure to stress and glucocorticoids (GC) can redirect the cell fate of differentiating neural progenitor cells toward oligodendrogenesis in the dentate gyrus of the adult rat rodent hippocampus [50]. Similar to neurogenesis, the generation of OPCs from neural stem/progenitor cells continues throughout adulthood. In the subcortical SVZ and in the hippocampal SGZ, anatomically different populations of stem cells retain the capacity to generate neurons, astrocytes and oligodendrocytes [51]. In this experimental study aimed at inducing significant changes, we show that exposure to six intense blast-waves affects the generation of neuronal lineage cells as well as oligodendrocyte development.
Materials and Methods
I Animals
The University at Buffalo Institutional Animal Care and Use Committee as outlined in HER05080Y and PTH10081Y protocols approved all animal procedures. Twenty-two adult, 3-4 months old, male C57BL/6J mice (Jackson Laboratory) used in the study were housed in the University at Buffalo Laboratory Animal Facility under standard conditions. Mice were randomly assigned to four groups: short-term control, short-term blast, long-term control or long-term blast.
II Blast-Wave Exposure
All mice were anaesthetized using ketamine (100 mg/kg) and xylazine (10 mg/kg). Animals in the blast groups were exposed to six blast waves with a peak intensity of approximately 185-peak dB SPL using equipment and procedures described in detail in our previous publication [52]. The blasts were presented once every five minutes. Six blasts were used in this experimental study in order to reduce between subject variability and enhance the likelihood of identify well-defined brain changes. Each anaesthetized mouse was exposed individually in an acoustically transparent wire-mesh cage (4 cm L, 2.5 cm W, 3 cm H) located approximately 2.5 cm in front of the mouth of the blast tube exit port. The head of the mouse faced towards the exit ports at 0° azimuth. The intensity of the blast was measured with a high-pressure sensor (137a23ICP Pressure Sensor, PCB Piezotronics). The non-blast control groups underwent the same treatment, but without the blast.
After the blast or non-blast procedure, mice were given carprofen (5 mg/kg) and following recovery from anaesthesia, the mice were transported back to their housing cages. In experiment 1, short-term recovery, the mice were heart-perfused, and the brains harvested 5 days after the blast or non-blast procedure. In experiment 2, long-term recovery, mice were heart-perfused, and the brains harvested 21 days after the blast or non-blast procedure.
III Tissue Preparation
Mice were anaesthetized by an intraperitoneal injection of ketamine/xylazine and sacrificed by transcardial perfusion with 4% paraformaldehyde. The brains were dissected and placed in 4% paraformaldehyde for 2 hours. The brains were cryopreserved by placing them into a sucrose solution of increasing concentrations (day 1 - 7.5% sucrose, day 2 - 15% sucrose, day 3 - 30% sucrose). Upon achieving density equilibrium, the brain was washed in 1X phosphate buffered saline (PBS), and the cerebellum was dissected. The tissue was then frozen in liquid nitrogen and stored at -80 oC until sectioning.
Brains were sectioned on a cryostat (Microm HM 505 N) in the coronal plane at 40 µm thickness. The start and end points for each region of interest (ROI) in the study was determined using the Paxinos Mouse Atlas: for SVZ (bregma +1.18 mm - bregma -0.22 mm), SGZ (bregma -0.94 mm - bregma - 4.16 mm) and cortex (bregma +1.18 mm - bregma -0.22 mm). All sections were serially transferred into 48-well plates containing a mixture of anti-freezing solution in accordance to IHC World protocol. The prepared material yielded approximately 30 sections per ROI (SVZ, SGZ, and Cortex). The first tissue was selected at bregma 1.10 mm for both SVZ and Cortex; the first tissue was selected at bregma - 1.06 mm for SGZ. Thereafter every sixth section was selected for immunostaining with five sections per ROI (Cortex, SVZ, and SGZ).
IV Immunostaining
Sections were rehydrated in PBS for 10 minutes, post-fixed with 4% paraformaldehyde for 10 minutes than washed twice in 1X PBS (20 minutes) and permeabilized with 0.5% Triton-X-100 (15 minutes). Sections were blocked with protein block (BioGenex) (1 hour at room temperature) and then incubated overnight at 4 °C with primary antibody of choice (Table 1) diluted in Antibody Diluent (IHC World cat # IW-1000). Subsequently, the sections were incubated for 2 hours at room temperature with secondary antibody also diluted in Antibody Diluent (IHC World). For double staining, secondary staining followed immediately after the primary staining according to the mentioned above. Slides were cover-slipped with Fluoromount II with DAPI (Cat. # 17985-50, Electron Microscopy Sciences) for nuclei visualization.
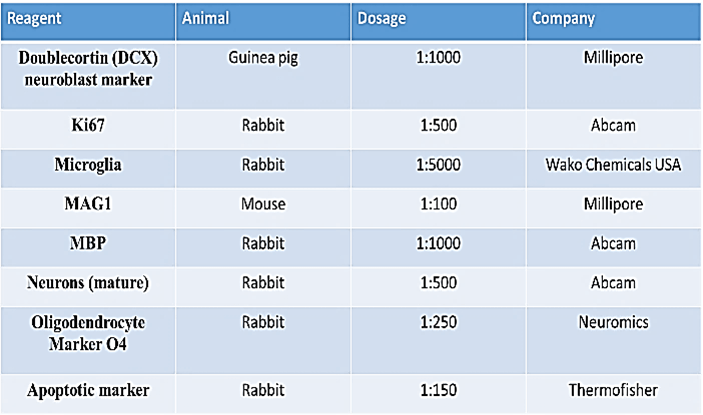
The primary antibodies used were guinea pig anti-DCX (AB2253, Millipore), rabbit anti-Ki67 (ab15580, Abcam), rabbit anti-microglia (NCNP24, Wako Chemicals USA), mouse anti-MAG1 (MAB1567, Millipore), rabbit anti-MBP (ab40390, Abcam), rabbit anti-Pan NeuN (ab104225, Abcam), rabbit anti-O4 (MO15002, Neuromics), rabbit anti-caspase3/7 (Asp175, Thermo Fisher). The following secondary antibodies were used: goat anti-guinea pig IgG H&L Cy5.5 (ab6967, Abcam), Alexa Fluor® 568 goat anti-rabbit IgG (A11011, Life Technologies-Molecular Probes), anti-MAG clone 513 directly conjugated to Alexa Fluor® 488, Alexa Fluor® goat pAb to mouse IgM (ab150121, Abcam).
V Image Analysis
Non-stereological imaging was performed using a Zeiss Axio Imager upright fluorescence microscope equipped with Zen Blue 2.3 software. Stereological analysis was performed in the Stereology Laboratory of the Western NY Stem Cell Culture and Analysis Center using an Olympus BX61 fully automated microscope equipped with a UIS2 optical system. Analyses were performed using newCAST software (Visiopharm, Denmark). Regions of interests (ROI) were outlined or masked using the Paxinos mouse brain atlas as a reference. A subset of ROI was then sampled using Optical Fractionator. Regions were randomly sampled at equidistant fields of vision (FOV). The areas sampled, (depending on density per area of events), were as follows: 55% of SVZ, 45% of SGZ and 25% of the cortex.
The FOVs were viewed at a set area-sampling fraction (ASF) of the masks using newCAST sampling software at 60X magnification for all areas. FOVs consisted of stacks of images taken through the z-axis of the ROI at a set interval (step size) of 2 micrometers for all areas. Care was taken to ensure that the entire depth of the section was imaged. The emission wavelength was set in the following order while digitizing the tissues – 603 nm, 530 nm, and 461 nm. Estimates per animal group were analysed for statistically significant differences. Statistical analysis was performed using SPSS (IBM and GraphPad Prism) software. Control and blast results were compared in short-term and in long-term experiments using unpaired t-test. Differences were considered significant with p ≤ 0.05.
Results
I Blast Induced Activation of Microglia and Astroglia
To determine whether the blast wave exposure affected microglia, the brain tissue was stained for the allograft inflammatory factor 1, also known as ionized calcium-binding adapter molecule 1 (IBA1), which labels activated microglial cells. In cortical regions of control brain sections, we observed the presence of small cells with short processes weakly immunolabeled by the IBA1 antibody. A representative image from a control mouse is shown in (Figure 1A, left panel). In contrast, strong IBA1 staining occurred in the brains of blast-exposed mice (Figure 1A, right panel). The morphology of most microglia in control brains was characterized by a small soma with short processes whereas the majority of microglia in the blast-exposed brains had rounded somas with elongated processes or M2-type microglia morphology (Figure 1A). The total spread of microglial processes was measured using ImageJ as illustrated in (Figure 1B). In cortical region of the blast wave exposed mice, the mean length of the microglial processes was significantly greater than that of control mice (Figure 1C). In addition to the M2-like cells, in the injured tissue, a few microglial cells displayed activated M1-type amoeboid bodies with ramified processes as described previously [53-57]. These results clearly show microglial activation in the blast-exposed brain cortex.

Figure 1: Blast induced neurotrauma. A) Activated microglia in the cortex. IBA1 - white (secondary Alexa 568). Microglia staining in Non-blast (A) and Blast (B). Magnification – 20X. B) The span of the process was measured using ImageJ software. Measuring guidelines were established. Straight line was extended from the hillock of the cell to the most extended point of the process. The span was determined from base of processes to the most extending point of process and calculated in the form of percentage microglial span shown on (Figure 1C). C) Cell density and length/spread of processes in the cortical region of ST control and blast mice brains. There was a significance increase in the average total spread of microglial processes in ST blast as compared to its control. D) Blast induced astrocytosis in cingulate cortex, magnification 10X - staining with anti-GFAP and secondary antibody with Alexa 568 (red).
Brain injuries and neuronal damage are typically accompanied by astrocytosis. To determine whether astrocytes became activated in our blast paradigm, we stained brain sections for GFAP, a protein highly expressed by reactive astrocytes. We found few reactive astrocytes in the cingulate and in the dorso-lateral cortical region of the control mice. In contrast, in the blast-exposed mice, heavily GFAP-stained astrocytes were observed throughout the cortex 5 days post-blast (Figure 1D). Thus, the blast exposure lead to a rapid microglial and astrocytic responses.
II Blast Induced Damage and Regrowth of Myelinated Axons
To identify potential injuries that could underlie the microglia and astroglia activations, we analysed the effects of blast on cortical myelinated axons. Staining with Black Gold II revealed dense myelinated fibers in the superficial cortical zone (zone I on Figure 2A); many of these were organized in horizontal bundles, and vertically organized bundles extending mainly between subcortical zones III and II and to a lesser extent zone I. At 5 days post-blast, we observed a marked loss of Black Gold II-stained fibers in all three zones of the medial cingulate cortex (Figure 2A) as well as the dorsolateral motor cortex (Figure 2B).
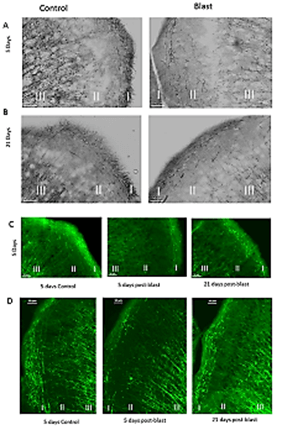
Figure 2: Blast induced loss and reappearance of cortical myelinated fibers. A) Cingulate Cortex and B) Dorso-lateral motor Cortex. Black Gold II staining – myelin fibers are stained black. Magnification – 20X. Cortical zone (I) and deeper subcortical zones (II and III) are marked. C) Loss (5 days) and reappearance (21 days) of myelin fibers in the cingulate cortex. Myelin associated glycoprotein staining (green). Magnification- 10X. D) Loss of myelin fibers in zone I and II in 5 days post blast (compare to Figure 5D). control and disorganized myelin fibers at 21 days after blast in zone I (cortex) and subcortical layers II, respectively Myelin basic protein (MBP) staining (red) Magnification – 20X.
We immunostained brain section for myelin-associated glycoprotein 1 (MAG1) at 5 days and 21 days post-blast and compared them to their controls. In controls, the cingulate cortex displayed a characteristic pattern of normal myelination with myelinated fibers extending from subcortical zone III to subcortical zone II and cortical zone I; uniform short fibers were distributed in cortical zone I (Figure 2C). Similar to the Black Gold II staining pattern, at 5 days post-blast there was a marked loss of the MAG1+ fibers in all three zones including the loss of the myelinated fibers extending from zone III to zones II and I (Figure 2C). The changes observed 5 days post-blast were followed by reappearance of myelinated processes in layer I and II at 21 days post-blast (Figure 2D). However, the reappearance of myelinated fibers in zone II (Figure 2D), lacked the predominantly vertical (perpendicular to the cortical surface) directionality characteristic of the control mice (Figures 2A-2C). Taken together, these observations suggest that the blast had temporarily damaged cortical and subcortical myelinated axons, but that by 21 days post-blast, the myelination has been partially restored.
III Blast Induced Apoptosis
To determine if the blast exposure lead to cell death, we stained brain sections with anti-caspase 3/7 antibody, which identifies apoptotic cells. In the cingulate cortex 5 days post-blast, we observed widespread, strong staining of caspase 3/7+ cells (Figure 3A2) compared to weak caspase 3/7+ staining in control mice (Figure 3A1). A marked reduction in the number of caspase 3/7+ cells was observed 21 days post-blast (Figure 3A3). These results indicate that there is a transient period of programmed cells death in cingulate cortex during the first week post-blast that has largely resolved itself by three-week post-blast.
Blast-induced apoptosis was not limited to superficial cortical regions but was observed also in the deeper sub-ventricular zone (SVZ), containing a stem cell niche (Figure 3B). To investigate the relationship between programmed cell death and neurogenesis, brain sections were co-stained for caspase 3/7+ and DCX, which labels developing neuroblasts. At 5 days post-blast, we observed intense caspase 3/7+ staining of SVZ cells many of which were also positive for DCX. Examples of single-stained SVZ caspase 3/7+ cells and double-stained DCX/caspase 3/7+ apoptotic neuroblasts are shown in (Figure 3B); double-stained cells of this type were absent in the SVZ of control mice. These results indicate the blast exposure induced apoptosis in developing neuroblasts as well as other cells within the SVZ.
To determine if cell death extended to hippocampal cortex and affected developing neurons in the dentate gyrus SGZ, we analysed caspase3/7 expression in the hippocampus in blast-exposed and control mice. Caspase3/7 labeling was largely absent in the hippocampus of control and blast-exposed mice (Figures 3C-3F); however, many DCX+ cells were present in the dentate gyrus of both controls and blast-exposed mice (note blue arrows pointing to DCX+ cells in Figure 3C-3F).
While caspase3/7 was absent from the CA 2/3 area of the hippocampus of control and blast-exposed mice at 5 days and 21 days post-treatment, there was extensive DCX labeling in CA 2/3. DCX labeling appeared to be greater in the blast-exposed mice compared to controls suggesting that the blast exposure increased neurogenesis. The blue arrows on (Figure 3E) point to the characteristic pair of post-mitotic DCX+ cells in CA 2/3 after blast. At 21 days post-blast, some of the DCX+ cells displayed extended processes indicating their further neuronal differentiation and possible integration into the hippocampal circuit.
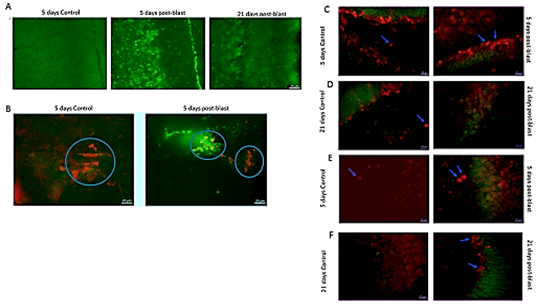
Figure 3: Blast induced apoptosis throughout brain. A) Apoptosis in cingulate cortex. Caspase3/7/– Green. Magnification 20X. B) Distribution of immature (DCX+, red) and apoptotic (caspase3/7/, green) cells in SVZ region 5 days post blast exposure. Magnification – 40X. Note apoptotic (green) cells and double stained (yellow) apoptotic neuroblasts cells in blast SVZ. C-F) Absence of apoptotic cells in hippocampus (caspase3/7 green stain), and presence of DCX+ cells (red stain, examples indicated with blue arrows) in the hippocampus dentate gyrus (C & D) and in CA2/3 area (E & F). Magnification 40X.
IV Effect of Blast on DCX+ Neuroblasts/Immature Neurons Cells in SVZ, Cingulate Cortex and Hippocampus
To determine whether the blasts affected early neuronal development, we performed stereological counting of DCX+ neuroblasts. In the SVZ, we found no significant effect of blast on the total numbers of DCX+\ cells (Figure 4A) at 5 days post-blast. However, at 21 days post-blast, there was a significant 2-fold increase in DCX+ cell density in the SVZ of the blast-exposed mice compared to control mice indicating an increased neurogenesis (Figure 4B). In the cingulate cortex, we found no blast- induced changes in the DCX+ population at the 5- or 21- days recovery times (Figures 4C & 4D). In contrast, stereological counting in the SGZ of the hippocampal dentate gyrus revealed an increase in the DCX+ cell population, which attained statistical significance 21-days post-blast (Figures 4D & 4E).
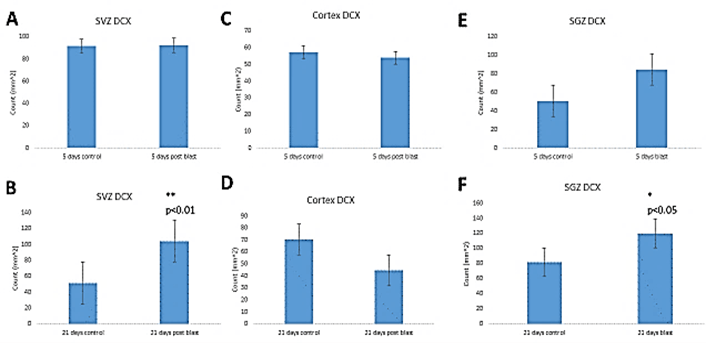
Figure 4: Stereological quantification of Doublecortin (DCX+) cells. A & B) Sub-ventricular zone – (A) short term recovery (5 days, ST); (B) – long term recovery (21 days, LT); C & D) cingulate cortex – (C) short term recovery (5 days, ST) (D) – long term recovery (21 days, LT); E & F) hippocampal dentate gyrus Sub-granular zone (SGZ)– (E) short term recovery (5 days, ST), (F) – long term recovery (21 days, LT).
V Effect of Blast on Oligodendrogenesis
Given the blast-induced loss of myelin in the cortex at 5 days post-blast followed by reappearance of myelinated fibers at 21 days post-blast (Figure 1), we studied the changes in oligodendrocytic cells at 5 days and 21 days post-blast. In order to observe myelin along with oligodendrocyte progenitors, we co-immunostained brain sections with an MBP antibody (green stain) and anti-Olig 2 (O2) antibody (red stain), which stains Olig 2, a protein expressed during early determinant of oligodendrocyte fate specification (Figure 5). At 5 days post-blast, the loss of cortical MBP+ fibers were accompanied by a widespread appearance of cortical O2+ cells (Figures 5A1 vs 5A2). Because O2+ cells only seldom seen in the cortex of control mice, these results suggested that the blast exposure activated cortical oligodendrogenesis. These early changes were followed by the reappearance of the myelinated fibers at 21 days, post-blast (Figure 2) and a decrease in the number of O2+ oligodendrocyte progenitor cells (Figure 5B3).
To determine if the changes in the O2+ cell population lead to oligodendrocyte development, we stained brain sections for O4, which is expressed by oligodendrocytes, but may also be present in developing OPCs. To observe the developmental progression of oligodendrocytes in the cingulate cortex, we also co-immunostained sections with an O2 antibody (red stain) and an O4 antibody (green stain). Figure 5 shows examples of O2+ cells in controls and from 5 days post-blast mice. In blast-exposed mice, the increase in O2+ cells illustrated in (Figure 5B1) (compared control in Figure 5B2) was accompanied by reduced O4 staining. However, at 21 days post-exposure, many O4+ cells were present in blast-exposed animal compared to controls suggesting an increased maturation of the O2+ progenitors (Figures 5B2 vs 5B3).
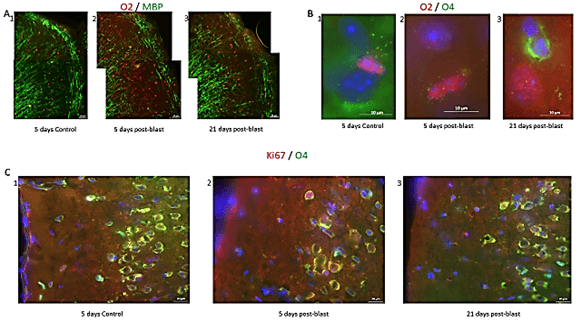
Figure 5: Blast induced effect on myelin and oligodendrocytic cell lineage. A) Loss and reappearance of myelin fibers and oligodendrocytes in the cortex. Myelin (is this MBP or another antibody) fibers stained green and O2 oligodendrocytes stained red. Magnification – 20X. B) Examples of O2+ (red), O4+ (green), DAPI (blue) cells in the cingulate cortex in 1) control and in 2) blast, ST recovery, tissues, showing O2+ cells. 3) Example of O4+ cell (green) in cingulate cortex blast, LT recovery. Magnification – 100X. C) Examples of Ki67+ (red), O4+ (green), DAPI (blue) cells in the cingulate cortex. In 1) control, ST condition, see higher O4+ cells while in 2) blast, ST recovery, loss of O4+ cells and increase in Ki67+ cells. 3) Blast, LT recovery, increase in O4+ cells toward control levels and increase in Ki67+ compared to control. Magnification – 40X.
To evaluate the blast-induced changes in oligodendrocytes, we performed stereological quantification of the O4+ cells in the cingulate cortex and SVZ, another potential source of cortical oligodendrocytes, as well as in the hippocampus where new oligodendrocytes are produced locally. To identify mitotically active OPC, we double-labeled cells for O4 and Ki67 (Figure 6). At 5 days post-blast, we found high numbers of O4+ cells in all brain areas and fewer OPC coexpressing Ki67 (Figure 5C1). Given the apparent loss of O4+ and O4+/Ki67+ cells that appeared to be followed by recovery, we quantified these cells using the stereology. At 5 days post-blast, the population of O4+ oligodendrocytes in the cingulate cortex was reduced three-fold compared to control (Figure 6A). However, at 21 days post-blast, the O4+ population recovered to levels seen in the control group (Figure 6B). This recovery was verified by the analysis of combined 5- and 21-day post-exposure results. Two-way ANOVA demonstrated an overall effect of blast (p<0.05) with a significant blast-induced loss of O4 cell at 5 days post-blast (p<0.05); in blast-exposed mice there was a significant difference between 5- and 21-days groups (p<0.05).
While there was a greater than two-fold increase in total numbers of Ki67+ cells at 5 days post-blast (p<0.01), the percentage of double-labeled cells was significantly reduced by 50%. These results together with the 2-fold loss of all O4+ cells amounted to a 4-fold decrease in proliferating O4+ oligodendrocytes. After 21 days, the fraction of proliferating (Ki67+ + O4+/O4+) was no longer significantly different from control (Figure 6B). In the SVZ, we found no blast induced changes in the numbers of Ki67+ or O4+ labeled cells at either 5- or 21- days post-blast. Approximately 20% of O4+represented proliferating Ki67+ cells and the observed reduction did not attain statistically significant levels. In the hippocampal SGZ, we found no blast-induced statistically significant changes in the O4+ or O4+ + Ki67+/O4+ cell counts.
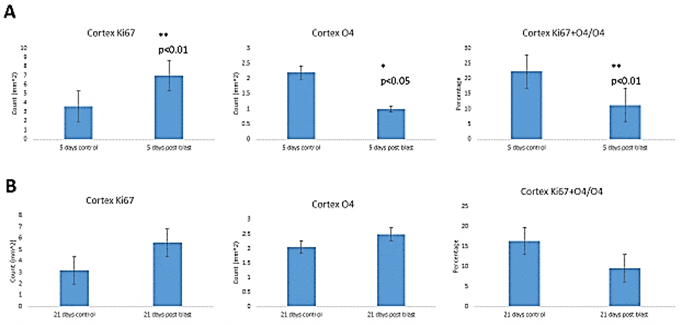
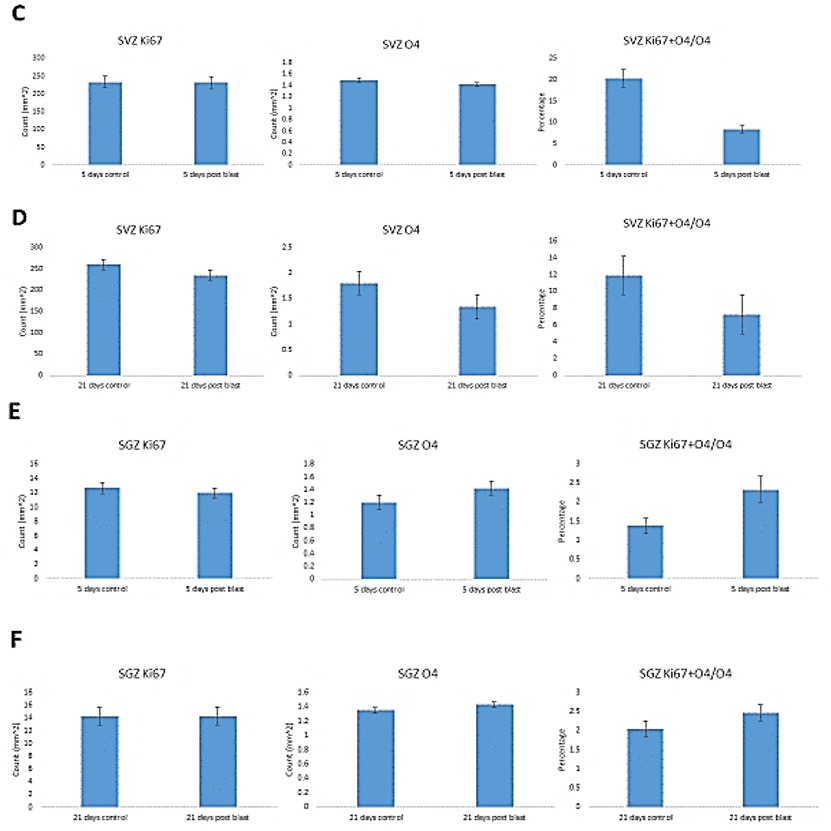
Figure 6: Stereological quantification of blast induced effects on O4, Ki67 cells and percentage of O4 cells that expressed Ki67. Cingulate cortex – A) short term (ST) and B) long term (LT) recovery. SVZ - C) short term and D) long-term recovery. SGZ – E) short term and F) long-term recovery.
Discussion
These results show that a series of six acoustic blasts of ~185 dB peak SPL leads to an acute injury of neurons in the brain cortex and to cortical and subcortical cell death accompanied by an activation of the microglia and astroglia. These pathological changes were followed by increased neurogenesis in the SVZ and SGZ and by increased cortical oligodendrogenesis.
I Microglia
Microglia activation, the first line of defense for the brain, is sensitive to minute biochemical changes in the CNS and extracellular potassium [58, 59]. Depending on the extent of tissue damage, the microglial response ranges from small morphological changes to full-blown activation involving phagocytosis of cell debris [60]. After brain injury, the activated microglia may transit to the non-phagocytic, anti-inflammatory, neuroprotective M2 type or to the neurotoxic M1 type [53-57]. M1 microglial cells have an amoeboid morphology, short processes and phagocytic, pro-inflammatory and neurotoxic properties.
In response to the blasts, we observed changes in microglia characterized by a round soma with elongated processes resembling M2-type microglia. The increase in M2-type cells may be involved in anti-inflammatory responses, tissue repair and remodeling as found in other kinds of brain injuries [61]. The cells could migrate towards the site of the blast-induced injury, clear apoptotic cells and cellular debris creating an initial neuro-protective state. Relatively few M1-like microglial cells with enlarged, amoeboid soma and thick processes were observed post-blast. The appearance of M1-like cells could produce toxic cytokines thereby damaging surrounding neuronal processes, neurons and oligodendrocytes [61].
II Astroglia and GFAP
Our blast wave exposure induced extensive astrogliosis manifested by a widespread appearance of reactive astrocytes 5 days post-blast. Astrocytes are involved in modulating the blood brain barrier, neuronal circuits and synapse reorganization, and inflammatory response [62, 63]. During acute brain injury, astrocyte activation may be neuroprotective although chronic activation has been linked to neurodegeneration [64, 65]. Our results are consistent with those of Cernak et al., who found that mice subjected to 183 kPa, or 213 kPa, shock waves had a nearly four-fold increase in the levels of GFAP in both hippocampus and brainstem 1 day post-shock and GFAP remained elevated out to 30 days [66]. In addition, rats subjected to 10 and 17 psi blast wave exhibited a 60% increase in GFAP protein.
III Apoptosis
These microglia and astrocytotic responses were accompanied by a widespread, transient burst of programmed cell death within the cortex and SVZ. These findings concur with previously demonstrated studies, which showed increased levels of caspase-3 up to 3 weeks post blast wave [67, 68]. Apoptosis in cortical oligodendrocytes has been shown to reach a peak 2 days post-blast in the external capsule and 7 days post-blast in the corpus callosum and fimbriae following a cortical impact injury [27]. In the SVZ, apoptosis was associated with DCX+ neuroblasts consistent with earlier findings showing that the SVZ neuronal progeny are vulnerable to trauma such as hypoxia-induced ischaemia while the stem cells were more resilient [69]. We found no apoptotic cells in the hippocampus, indicating that this area was spared from the blast-induced damage. Our results differ from the intense hippocampal apoptosis following global ischaemia and concussion [70, 71].
IV Demyelination and Remyelination
The blast-induced early loss of myelinated cortical and subcortical axons is indicative of transient demyelination of the surviving axons but also degeneration of myelinated axons. While remyelination was observed in the 21 days post-blast group, it re-appeared in a disorganized fashion, i.e., lacked the typical radial orientation. The timeline with which the repair occurs falls in line with previous results, which have shown endogenous murine precursors would likely completely repair a demyelinated lesion in 3-4 weeks [72]. The reappearance of such misdirected myelinated fibers suggest that the original cortical axons had degenerated but had subsequently regrown. The myelin sheath could have been lost or diminished as a result of the insult to the myelinating cells as reported previously [73]. In the central nervous system, myelin, a lipid-rich, multi-lamellar membrane is produced by oligodendrocytes, which wraps around axons. A single oligodendrocyte has the ability to myelinate several axonal internodes and their loss has been shown to block signal propagation within neuronal networks [74, 75]. In ischaemia, demyelination appears to be a result of a damage to the oligodendrocytes [76-78]. In our investigation, the loss and the reappearance of myelin after the blast could also reflect an initial loss of the oligodendrocyte population and their subsequent regeneration.
V Neurogenesis
Our blast exposure did not affect the global population of cortical neuroblasts; however, an increase in the neuroblast population in the SVZ was found. This increase developed gradually during the 21 days post-blast recovery period. This increase shows a neurogenic response in the area that supplies new neurons to the olfactory bulb (OB) as well as into the cortex. It is conceivable that these new cells arose through increased differentiation of the existing pool of post-mitotic progenitors because no increase in SVZ cell proliferation (Ki67 labelling) was found at either 5- or 21-days post-blast. In the quiescent state, the SVZ-derived NPCs are responsible for the generation of neuroblasts that migrate to the OB through the rostral migratory stream (RMS) [79-81]. However, when stimulated by an injury or reactivation of developmental Integrative Nuclear FGFR1 Signaling, the SVZ NPC also produce cortical and subcortical neurons [82, 83]. Since the population of DCX+ cells did not increase in the cortex, follow up studies are needed to determine whether progeny of the SVZ DCX+ cells have actually migrated into the cortex and differentiated into DCX+ expressing neurons.
In the SGZ, the blast triggered an increase in DCX+ neuroblasts, which could lead to an increase in hippocampal neurons similar to the increase observed in the hippocampus after hypoxia-ischaemia [84]. Similar to the SVZ, the addition of DCX+ neuroblasts in the dentate gyrus was not derived through increased proliferation of the SGZ cells because no changes were found in Ki67+ cells at either 5- or 21-days post-blast. One interpretation of these results is that the blast-induced increases in SGZ neuroblasts population occurred by increased neuronal lineage differentiation of the SGZ transit amplifying progenitors. The molecular mechanisms triggering an increase in SVZ and SGZ neuroblasts may be due in part to microglial activation. M2 microglia are known to increase the development and survival of neurons and microglia-secreted insulin-like growth factor (IGF-1) and brain derived neurotrophic factor (BDNF) which promote neuronal development and differentiation [85-87]. The increases in the number of DCX+ cells specifically in the lateral ventricle and in the dentate gyrus likely predicts the increased formation of neurons as indicated in several brain injury publications [49, 88-95].
VI Oligodendrocytes
Oligodendrocytes have the highest metabolic rates amongst all brain cells because of the metabolic demands needed to produce and maintain myelin [96, 97]. Their respiration rate is twice as high as neurons making oligodendrocytes particularly prone to the damage by hypoxia [98]. Oligodendrocytes also have low levels of glutathione, a major antioxidant and, therefore, are especially susceptible to the oxidative stress [99]. Activation of microglia and the release of reactive oxygen and nitrogen species have been linked to injury of both oligodendrocytes and neurons [100].
Interestingly, in our blast exposure activated microglia and astrocytes; this activation was correlated with the loss of oligodendrocytes and neuronal injury. The degeneration of cortical MAG1+ fibers and the loss of O4 oligodendrocytes (~65%) at 5 days post-blast could be due to the appearance and action of the M1 microglia. However, already at that time point, we observed the widespread, transient appearance of cortical O2+ cells in which microglia could also play a role. Specifically, the M2-type microglia, which predominated in our blast-injured brain tissue, are known to enhance oligodendrocyte differentiation [101]. Following a TBI, the loss of oligodendrocytes via apoptosis was associated with an increase in newborn O2+ cells, which reached a peak 7 days after TBI [27]. Hence, the increase in O2 cells observed in our study could be triggered by the blast-induced loss of the mature oligodendrocytes.
At 5 days post-blast, we observed an overall increase in the number of proliferating cortical cells. Since the fraction of Ki67+ cells co-expressing O4 was markedly reduced (~79%), this would suggest that no OPC had been generated at this time. However, at 21 days post-blast, O2+ oligodendrocytic progenitors had decreased while Ki67+/ O4+ OPC had recovered and mature oligodendrocytes were restored to control levels. Thus, we propose that during post-blast remyelination there was a sequential upregulation of oligodendrocytic progenitors (O2), which differentiated to OPC (Ki67/O4) and in turn produced the mature (O4) oligodendrocytes. This sequence is consistent with the findings based on genetic cell tracing [102]. During remyelination in PDGFRα-Cre ERT2 mice, the oligodendrocyte density increased at the expense of O2+ progenitor cells indicating that progenitors had developed into myelinating oligodendrocytes [102]. The increased production of oligodendrocytes from OPCs has been reported in experimental models where focal demyelination occurs adjacent to the SVZ and in postmortem tissue obtained from MS patients [103, 104].
During development, forebrain oligodendrocytes have been shown to first originate from OPCs in the embryonic SVZ of the ventral telencephalon [105]. The early waves of OPCs are transitory and superseded by OPCs originating latter from dorsal telencephalic sources [105, 106]. In adult mice, OPCs make up 8-9% of the cell population in the white matter and 2%-3% of the population in the gray matter [107, 108]. Many OPCs divide to form mature myelinating oligodendrocytes, but others linger as slowly dividing immature cells, which are widely distributed within the adult brain [32, 33, 104, 109]. It has been proposed that cortical gray and white matter might be myelinated by different subpopulations of OPCs [110]. One-group of cells migrating from the SVZ are thought give rise to the oligodendrocytes that carry out myelination during development, while another group gives rise to backup OPC that reside in the cortex and contribute to damage-induced remyelination.
In the adult brain, SVZ progenitors have been shown to generate new oligodendrocytes after demyelinating lesions of the corpus callosum (CC) [32, 109, 111, 112]. In contrast, in our investigation we observed no changes in OPC or O4 oligodendrocytes in either the SVZ or SGZ. Thus, the newly generated cortical oligodendrocytes observed after our blast exposure did not come from these stem cell niches. Instead, our findings are consistent with a local regenerative model in which cortical O4 cells are resupplied within the cortex. The loss and reappearance of cortical myelinating cells has been reported following mechanical brain injury where oligodendrocytes were depleted in the ipsilateral cortex and external capsule at 7 days after the initial trauma and returned to control levels at 5 weeks [27].
A major unsolved problem with insult-induced neurogenesis and oligodendrogenesis studies is whether these processes lead to functional recovery and reversal of neurological deficits [113, 114]. Even if adequately myelinated, the newly generated, but miss-wired neurons could distort the normal cortical circuitry adding to long-term deficits produced by the blast injury. Therefore, the assimilation of newly generated neurons into the brain circuitry needs to be evaluated both anatomically and functionally since these newly added cells and axons could remain immature and/or lead to the miss-wiring of the brain [115].
VII Synopsis
We have shown a significant increase in neuroblasts in the sub-ventricular zone and hippocampus of adult mice three weeks after blast wave exposure. It remains to be determined whether: 1) the SVZ-produced neurons migrate to the site of injury and integrate into the cortical circuits and 2) what effect the SGZ generated neurons have on hippocampal function. Blast wave exposure triggered an initial axonal loss and demyelination in cortex which was followed by axonal regrowth and remyelination of cortical axons. Because axonal regrowth was still misdirected at 21 days post-blast, further research is needed to determine if cortical function will return to normal with longer recovery times.
Article Info
Article Type
Research ArticlePublication history
Received: Fri 10, Jul 2020Accepted: Wed 29, Jul 2020
Published: Fri 18, Dec 2020
Copyright
© 2023 Michal K. Stachowiak. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.NNB.2020.03.07
Figures & Tables
References
- Dale R (2006) Guerila Warfare and Counterinsurgency. J Conflict Studies 24.
- Taddeo V (2012) U.S. Response to Terrorism: A Strategic Analysis of the Afghanistan Campaign. J Strategic Security 3: 27-36.
- Wang H, Zhang YP, Cai J, Shields LB, Tuchek CA et al. (2016) A Compact Blast-Induced Traumatic Brain Injury Model in Mice. J Neuropathol Exp Neurol 75: 183-196. [Crossref]
- DoD worldwide numbers for TBI (2018) Defense and Veterans Brain Injury Center.
- Ling GS, Ecklund JM (2011) Traumatic brain injury in modern war. Curr Opin Anaesthesiol 24: 124-130. [Crossref]
- Vasterling JJ, Brailey K, Proctor SP, Kane R, Heeren T et al. (2012) Neuropsychological outcomes of mild traumatic brain injury, post-traumatic stress disorder and depression in Iraq-deployed US Army soldiers. Br J Psychiatry 201: 186-192. [Crossref]
- McKee AC, Robinson ME (2014) Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement 10: S242-S253. [Crossref]
- Snell FI, Halter MJ (2010) A signature wound of war: mild traumatic brain injury. J Psychosoc Nurs Ment Health Serv 48: 22-28. [Crossref]
- Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R et al. (2011) Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. J Neurotrauma 28: 1101-1119. [Crossref]
- Chen YC, Smith DH, Meaney DF (2009) In-vitro approaches for studying blast-induced traumatic brain injury. J Neurotrauma 26: 861-876. [Crossref]
- DePalma RG, Burris DG, Champion HR, Hodgson MJ (2005) Blast injuries. N Engl J Med 352: 1335-1342. [Crossref]
- Ritenour AE, Baskin TW (2008) Primary blast injury: update on diagnosis and treatment. Crit Care Med 36: S311-S317. [Crossref]
- Elder GA, Mitsis EM, Ahlers ST, Cristian A (2010) Blast-induced Mild Traumatic Brain Injury. Psychiatr Clin North Am 33: 757-781. [Crossref]
- Taylor PA, Ludwigsen JS, Ford CC (2014) Investigation of blast-induced traumatic brain injury. Brain Inj 28: 879-895. [Crossref]
- Cernak I, Wang Z, Jiang J, Bian X, Savic J (2001) Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma 50: 695-706. [Crossref]
- Chavko M, Watanabe T, Adeeb S, Lankasky J, Ahlers ST et al. (2011) Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J Neurosci Methods 195: 61-66. [Crossref]
- Hue CD, Cao S, Haider SF, Vo KV, Effgen GB et al. (2013) Blood-brain barrier dysfunction after primary blast injury in vitro. J Neurotrauma 30: 1652-1663. [Crossref]
- Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR et al. (2010) Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res 88: 3530-3539. [Crossref]
- Gu M, Kawoos U, McCarron R, Chavko M (2017) Protection against Blast-Induced Traumatic Brain Injury by Increase in Brain Volume. Biomed Res Int 2017: 2075463. [Crossref]
- Zhang Y, Yang Y, Tang H, Sun W, Xiong X et al. (2014) Hyperbaric oxygen therapy ameliorates local brain metabolism, brain edema and inflammatory response in a blast-induced traumatic brain injury model in rabbits. Neurochem Res 39: 950-960. [Crossref]
- Goldstein LE, Fisher AM, Tagge CA, Zhang X, Velisek L et al. (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4: 134ra60. [Crossref]
- Arun P, Abu Taleb R, Oguntayo S, Wang Y, Valiyaveettil M et al. (2013) Acute mitochondrial dysfunction after blast exposure: potential role of mitochondrial glutamate oxaloacetate transaminase. J Neurotrauma 30: 1645-1651. [Crossref]
- Sajja VS, Galloway MP, Ghoddoussi F, Thiruthalinathan D, Kepsel A et al. (2012) Blast-induced neurotrauma leads to neurochemical changes and neuronal degeneration in the rat hippocampus. NMR Biomed 25: 1331-1339. [Crossref]
- Arandjelovic S, Ravichandran KS (2015) Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16: 907-917. [Crossref]
- Jurewicz A, Matysiak M, Tybor K, Kilianek L, Raine CS et al. (2005) Tumour necrosis factor-induced death of adult human oligodendrocytes is mediated by apoptosis inducing factor. Brain 128: 2675-2688. [Crossref]
- Horiuchi M, Itoh A, Pleasure D, Itoh T (2006) MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendroglial progenitor cells. J Biol Chem 281: 20095-20106. [Crossref]
- Dent KA, Christie KJ, Bye N, Basrai HS, Turbic A et al. (2015) Oligodendrocyte birth and death following traumatic brain injury in adult mice. PLoS One 10: e0121541. [Crossref]
- Fok Seang J, Mathews GA, Ffrench Constant C, Trotter J, Fawcett JW (1995) Migration of oligodendrocyte precursors on astrocytes and meningeal cells. Dev Biol 171: 1-15. [Crossref]
- Doetsch F, Alvarez Buylla A (1996) Network of tangential pathways for neuronal migration in adult mammalian brain. Proc Natl Acad Sci U S A 93: 14895-14900. [Crossref]
- Doetsch F, Garcia Verdugo JM, Alvarez Buylla A (1997) Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17: 5046-5061. [Crossref]
- Doetsch F, Caille I, Lim DA, Garcia Verdugo JM, Alvarez Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97: 703-716. [Crossref]
- Gonzalez Perez O, Alvarez Buylla A (2011) Oligodendrogenesis in the subventricular zone and the role of epidermal growth factor. Brain Res Rev 67: 147-156. [Crossref]
- Menn B, Garcia Verdugo JM, Yaschine C, Gonzalez Perez O, Rowitch D et al. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci 26: 7907-7918. [Crossref]
- Altman J (1969) Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J Comp Neurol 137: 433-457. [Crossref]
- Lois C, Alvarez Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264: 1145-1148. [Crossref]
- Pencea V, Bingaman KD, Freedman LJ, Luskin MB (2001) Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol 172: 1-16. [Crossref]
- Kornack DR, Rakic P (2001) The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc Natl Acad Sci U S A 98: 4752-4757. [Crossref]
- Doetsch F, Garcia Verdugo JM, Alvarez Buylla A (1999) Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci U S A 96: 11619-11624. [Crossref]
- Stachowiak MK, Stachowiak EK (2016) Evidence-Based Theory for Integrated Genome Regulation of Ontogeny--An Unprecedented Role of Nuclear FGFR1 Signaling. J Cell Physiol 231: 1199-1218. [Crossref]
- Suzuki SO, Goldman JE (2003) Multiple cell populations in the early postnatal subventricular zone take distinct migratory pathways: a dynamic study of glial and neuronal progenitor migration. J Neurosci 23: 4240-4250. [Crossref]
- Seri B, Garcia Verdugo JM, Collado Morente L, McEwen BS, Alvarez Buylla A (2004) Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol 478: 359-378. [Crossref]
- Seri B, Garcia Verdugo JM, McEwen BS, Alvarez Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21: 7153-7160. [Crossref]
- Filippov V, Kronenberg G, Pivneva T, Reuter K, Steiner B et al. (2003) Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Mol Cell Neurosci 23: 373-382. [Crossref]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ et al. (2011) In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145: 1142-1155. [Crossref]
- Bonaguidi MA, Song J, Ming GL, Song H (2012) A unifying hypothesis on mammalian neural stem cell properties in the adult hippocampus. Curr Opin Neurobiol 22: 754-761. [Crossref]
- Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132: 645-660. [Crossref]
- Encinas JM, Vaahtokari A, Enikolopov G (2006) Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci U S A 103: 8233-8238. [Crossref]
- Encinas JM, Enikolopov G (2008) Identifying and quantitating neural stem and progenitor cells in the adult brain. Methods Cell Biol 85: 243-272. [Crossref]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B et al. (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23: 247-256. [Crossref]
- Chetty S, Friedman AR, Taravosh Lahn K, Kirby ED, Mirescu C et al. (2014) Stress and glucocorticoids promote oligodendrogenesis in the adult hippocampus. Mol Psychiatry 19: 1275-1283. [Crossref]
- Tognatta R, Miller RH (2016) Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology 110: 539-547. [Crossref]
- Newman AJ, Hayes SH, Rao AS, Allman BL, Manohar S et al. (2015) Low-cost blast wave generator for studies of hearing loss and brain injury: blast wave effects in closed spaces. J Neurosci Methods 242: 82-92. [Crossref]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K et al. (2007) Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab 27: 488-500. [Crossref]
- Thored P, Heldmann U, Gomes Leal W, Gisler R, Darsalia V et al. (2009) Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57: 835-849. [Crossref]
- Wang J, Yang Z, Liu C, Zhao Y, Chen Y (2013) Activated microglia provide a neuroprotective role by balancing glial cell-line derived neurotrophic factor and tumor necrosis factor-alpha secretion after subacute cerebral ischemia. Int J Mol Med 31: 172-178. [Crossref]
- Tang Y, Le W (2016) Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol 53: 1181-1194. [Crossref]
- Hu X, Li P, Guo Y, Wang H, Leak RK et al. (2012) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43: 3063-3070. [Crossref]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y et al. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8: 752-758. [Crossref]
- Gehrmann J, Matsumoto Y, Kreutzberg GW (1995) Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20: 269-287. [Crossref]
- Alvis Miranda HR, Alcalá Cerra G, Moscote Salazar LR (2013) Microglia: roles and rules in brain traumatic injury. Romanian Neurosurg 2013: 34-45.
- Streit WJ (2000) Microglial response to brain injury: a brief synopsis. Toxicol Pathol 28: 28-30. [Crossref]
- Bailey ZS, Grinter MB, VandeVord PJ (2016) Astrocyte Reactivity Following Blast Exposure Involves Aberrant Histone Acetylation. Front Mol Neurosci 9: 64. [Crossref]
- Sajja VS, Hlavac N, VandeVord PJ (2016) Role of Glia in Memory Deficits Following Traumatic Brain Injury: Biomarkers of Glia Dysfunction. Front Integr Neurosci 10: 7. [Crossref]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH et al. (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136: 28-42. [Crossref]
- Sajja VS, Hubbard WB, Hall CS, Ghoddoussi F, Galloway MP et al. (2015) Enduring deficits in memory and neuronal pathology after blast-induced traumatic brain injury. Sci Rep 5: 15075. [Crossref]
- Cernak I, Merkle AC, Koliatsos VE, Bilik JM, Luong QT et al. (2011) The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis 41: 538-551. [Crossref]
- Rubovitch V, Ten Bosch M, Zohar O, Harrison CR, Tempel Brami C et al. (2011) A mouse model of blast-induced mild traumatic brain injury. Exp Neurol 232: 280-289. [Crossref]
- Tompkins P, Tesiram Y, Lerner M, Gonzalez LP, Lightfoot S et al. (2013) Brain injury: neuro-inflammation, cognitive deficit, and magnetic resonance imaging in a model of blast induced traumatic brain injury. J Neurotrauma 30: 1888-1897. [Crossref]
- Romanko MJ, Rothstein RP, Levison SW (2004) Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J Cereb Blood Flow Metab 24: 814-825. [Crossref]
- Shimizu H, Ohgoh M, Ikeda M, Nishizawa Y, Ogura H (2007) Caspase-3-like protease activity-independent apoptosis at the onset of neuronal cell death in the gerbil hippocampus after global ischemia. Biol Pharm Bull 30: 1950-1953. [Crossref]
- Nakajima Y, Horiuchi Y, Kamata H, Yukawa M, Kuwabara M et al. (2010) Distinct time courses of secondary brain damage in the hippocampus following brain concussion and contusion in rats. Tohoku J Exp Med 221: 229-235. [Crossref]
- Dietz KC, Polanco JJ, Pol SU, Sim FJ (2016) Targeting human oligodendrocyte progenitors for myelin repair. Exp Neurol 283: 489-500. [Crossref]
- Maxwell WL (2013) Damage to myelin and oligodendrocytes: a role in chronic outcomes following traumatic brain injury? Brain Sci 3: 1374-1394. [Crossref]
- Hartline DK (2008) What is myelin? Neuron Glia Biol 4: 153-163. [Crossref]
- Hartline DK, Colman DR (2007) Rapid conduction and the evolution of giant axons and myelinated fibers. Current Biol 17: R29-R35. [Crossref]
- Uchida H , Fujita Y, Matsueda M, Umeda M, Matsuda S et al. (2010) Damage to neurons and oligodendrocytes in the hippocampal CA1 sector after transient focal ischemia in rats. Cell Mol Neurobiol 30: 1125-1134. [Crossref]
- Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y et al. (2000) Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 31: 1735-1743. [Crossref]
- Back SA, Craig A, Kayton RJ, Luo NL, Meshul CK et al. (2007) Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab 27: 334-347. [Crossref]
- Capilla Gonzalez V, Cebrian Silla A, Guerrero Cazares H, Garcia Verdugo JM, Quinones Hinojosa A (2013) The generation of oligodendroglial cells is preserved in the rostral migratory stream during aging. Front Cell Neurosci 7: 147. [Crossref]
- Sun W, Kim H, Moon Y (2010) Control of neuronal migration through rostral migration stream in mice. Anat Cell Biol 43: 269-279. [Crossref]
- van Strien ME, van den Berge SA, Hol EM (2011) Migrating neuroblasts in the adult human brain: a stream reduced to a trickle. Cell Res 21: 1523-1525. [Crossref]
- Narla ST, Klejbor I, Birkaya B, Lee Y, Morys J et al. (2013) Activation of developmental nuclear fibroblast growth factor receptor 1 signaling and neurogenesis in adult brain by alpha7 nicotinic receptor agonist. Stem Cells Transl Med 2: 776-788. [Crossref]
- Stachowiak EK, Roy I, Lee Y, Capacchietti M, Aletta JM et al. (2009) Targeting novel integrative nuclear FGFR1 signaling by nanoparticle-mediated gene transfer stimulates neurogenesis in the adult brain. Integr Biol (Camb) 1: 394-403. [Crossref]
- Miles DK, Kernie SG (2008) Hypoxic-ischemic brain injury activates early hippocampal stem/progenitor cells to replace vulnerable neuroblasts. Hippocampus 18: 793-806. [Crossref]
- Ziv Y, Schwartz M (2008) Immune-based regulation of adult neurogenesis: implications for learning and memory. Brain Behav Immun 22: 167-176. [Crossref]
- Morgan SC, Taylor DL, Pocock JM (2004) Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and delta-Notch signalling cascades. J Neurochem 90: 89-101. [Crossref]
- Schafer DP, Stevens B (2015) Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harb Perspect Biol 7: a020545. [Crossref]
- Balthazart J, Ball GF (2014) Doublecortin is a highly valuable endogenous marker of adult neurogenesis in canaries. Commentary on Vellema M et al. (2014): Evaluating the predictive value of doublecortin as a marker for adult neurogenesis in canaries (Serinus canaria) . J Comparative Neurol 522:1299-1315. Brain Behav Evol 84: 1-4. [Crossref]
- Vellema M, Ko MC, Frankl Vilches C, Gahr M (2014) What makes a marker a good marker?. Commentary on Balthazart J and Ball G (2014): Doublecortin is a highly valuable endogenous marker of adult neurogenesis in canaries. Brain Behav Evol 84:1-4. Brain Behav Evol 84: 5-7. [Crossref]
- Couillard Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M et al. (2005) Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21: 1-14. [Crossref]
- Karl C, Couillard Despres S, Prang P, Munding M, Kilb W et al. (2005) Neuronal precursor-specific activity of a human doublecortin regulatory sequence. J Neurochem 92: 264-282. [Crossref]
- Friocourt G, Kappeler C, Saillour Y, Fauchereau F, Rodriguez MS et al. (2005) Doublecortin interacts with the ubiquitin protease DFFRX, which associates with microtubules in neuronal processes. Mol Cell Neurosci 28: 153-164. [Crossref]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23: 257-271. [Crossref]
- Yang HK, Sundholm Peters NL, Goings GE, Walker AS, Hyland K et al. (2004) Distribution of doublecortin expressing cells near the lateral ventricles in the adult mouse brain. J Neurosci Res 76: 282-295. [Crossref]
- Spampanato J, Sullivan RK, Turpin FR, Bartlett PF, Sah P (2012) Properties of doublecortin expressing neurons in the adult mouse dentate gyrus. PLoS One 7: e41029. [Crossref]
- Morell P, Toews AD (1984) In Vivo Metabolism of Oligodendroglial Lipids. Boston, MA: Springer.
- Connor JR, Menzies SL (1996) Relationship of iron to oligodendrocytes and myelination. Glia 17: 83-93. [Crossref]
- Cammer W (1984) Carbonic anhydrase in oligodendrocytes and myelin in the central nervous system. Ann N Y Acad Sci 429: 494-497. [Crossref]
- Thorburne SK, Juurlink BH (1996) Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem 67: 1014-1022. [Crossref]
- Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI et al. (2003) Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol 62: 441-450. [Crossref]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM et al. (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16: 1211-1218. [Crossref]
- Zawadzka M, Rivers LE, Fancy SP, Zhao C, Tripathi R et al. (2010) CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 6: 578-590. [Crossref]
- Huang JK, Fancy SP, Zhao C, Rowitch DH, Ffrench Constant C et al. (2011) Myelin regeneration in multiple sclerosis: targeting endogenous stem cells. Neurotherapeutics 8: 650-658. [Crossref]
- Petratos S, Gonzales MF, Azari MF, Marriott M, Minichiello RA et al. (2004) Expression of the low-affinity neurotrophin receptor, p75(NTR), is upregulated by oligodendroglial progenitors adjacent to the subventricular zone in response to demyelination. Glia 48: 64-75. [Crossref]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M et al. (2006) Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9: 173-179. [Crossref]
- Chapman H, Waclaw RR, Pei Z, Nakafuku M, Campbell K (2013) The homeobox gene Gsx2 controls the timing of oligodendroglial fate specification in mouse lateral ganglionic eminence progenitors. Development 140: 2289-2298. [Crossref]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M (2008) Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci 28: 10434-10442. [Crossref]
- de Castro F, Bribian A, Ortega MC (2013) Regulation of oligodendrocyte precursor migration during development, in adulthood and in pathology. Cell Mol Life Sci 70: 4355-4368. [Crossref]
- Xing YL, Roth PT, Stratton JA, Chuang BH, Danne J et al. (2014) Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J Neurosci 34: 14128-14146. [Crossref]
- van Tilborg E, de Theije CGM, van Hal M, Wagenaar N, de Vries LS et al. (2018) Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 66: 221-238. [Crossref]
- Nait Oumesmar B, Decker L, Lachapelle F, Avellana Adalid V, Bachelin C et al. (1999) Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci 11: 4357-4366. [Crossref]
- Picard Riera N, Decker L, Delarasse C, Goude K, Nait Oumesmar B et al. (2002) Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci U S A 99: 13211-13216. [Crossref]
- Okano H, Sawamoto K (2008) Neural stem cells: involvement in adult neurogenesis and CNS repair. Philos Trans R Soc Lond B Biol Sci 363: 2111-2122. [Crossref]
- Okano H (2006) Adult neural stem cells and central nervous system repair. Ernst Schering Res Found Workshop 60: 215-228. [Crossref]
- Gage FH (2003) Brain, repair yourself. Scientific American 289: 46-53.