Presentation of A Cutaneous Granular Cell Tumor in A 52-Year-Old Man in Remote Australia
A B S T R A C T
Granular cell tumors are rare neural crest tumors which predominantly occur in the skin. While most of these tumors are benign, malignant potential exists. Malignant granular cell tumors behave aggressively and are associated with poor outcomes. However, the distinction can be difficult at initial diagnosis. This case describes the presentation of a benign cutaneous granular cell tumor to a remote health service in Northwest Queensland, Australia.
Keywords
Granular cell tumour, Rare skin lesions, Immunohistochemistry
Introduction
Skin is the largest organ of the body and while tumors of epithelial cells are most common, tumors of any of the cell lines can occur resulting in a diverse range of benign and malignant pathologies. Granular cell tumors are one such entity. The rarity of these tumors is illustrated in a 1980 paper which reviewed more than 410,000 specimens, with granular cell tumors only accounting for 0.03% of results [1]. These tumors are thought to be of neural cell origin, but nonneural granular cell tumors have also been identified [2-4]. Most granular cell tumors are benign, but a small proportion are malignant and behave aggressively with a median survival of 3 years [4]. Identifying the subset of malignant tumors within all granular cell tumors can be difficult and criteria have been developed to assist.
Case Description
A 52-year-old man was referred to Mt Isa Hospital with an expanding subcutaneous mass on his anterior right chest which had developed over the preceding two years. He had a past medical history of type 2 diabetes, and hypertension. Drainage had been attempted prior to referral with minimal fluid returned. Clinically this appeared as a 5 cm dermal mass which was firm and non-tender with no fixation to underlying anatomical structures. It had a 3 × 3 cm central protuberance with multiple hypopigmented punctae (Figure 1).
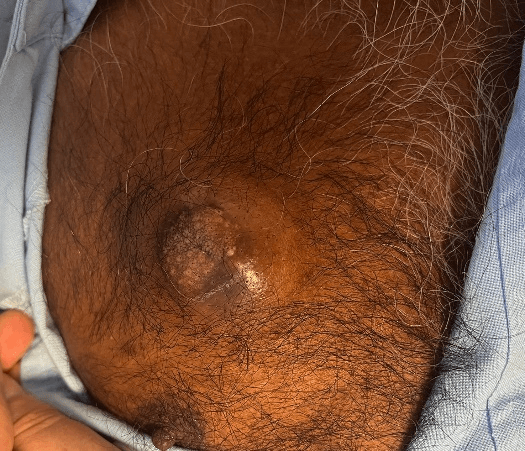
Figure 1: Dermal mass present on the right anterior chest wall which had developed over a 2-year period.
Computerised tomography (CT) and ultrasound (US) showed a 45 × 43 × 41 mm mass limited to the subcutaneous plane which did not extend into the underlying pectoralis major. Both modalities reported findings consistent with a sebaceous cyst (Figures 2 & 3). Clinical findings were not convincing of a sebaceous cyst but concerning for dermatofibrosarcoma protuberans, so a core biopsy was performed to aid diagnosis. Histology demonstrated a focal collection of polygonal cells with abundant granular eosinophilic cytoplasm and small, bland nuclei (Figure 4), which were positive for S100 (Figure 5), SOX10, and inhibin. These morphologic and immunohistochemical features were suggestive of granular cell tumor. He proceeded to excision with a wide margin and primary closure (Figures 6 & 7). Final histology confirmed the diagnosis of a granular cell tumor without high-risk features.
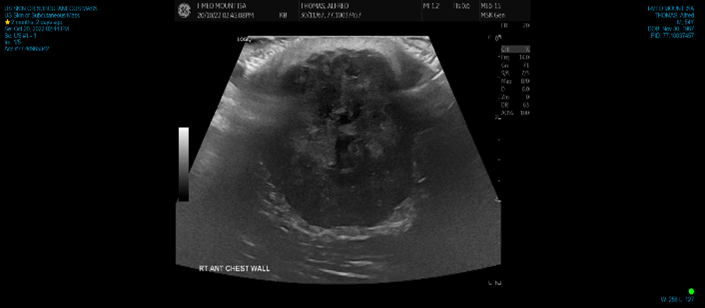
Figure 2: Ultrasound showing complex, avascular, hypoechoic mass along the right anterior chest wall limited to the subcutaneous plane and not extending into the underlying muscle.
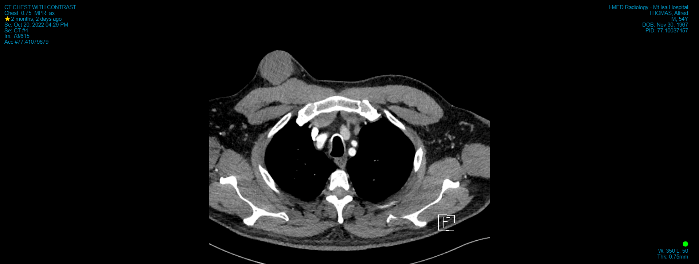
Figure 3: There is 4.5cm non-enhancing mass overlying the right pectoralis muscle.
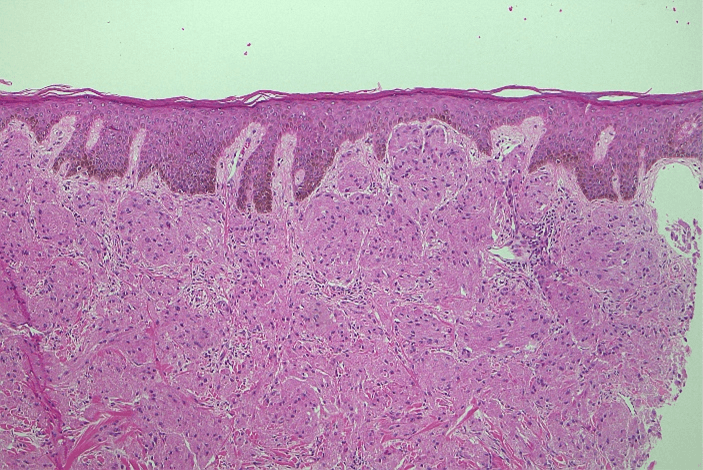
Figure 4: Hematoxylin and eosin stain showing focal collection of polygonal cells with abundant granular eosinophilic cytoplasm and small, bland nuclei.
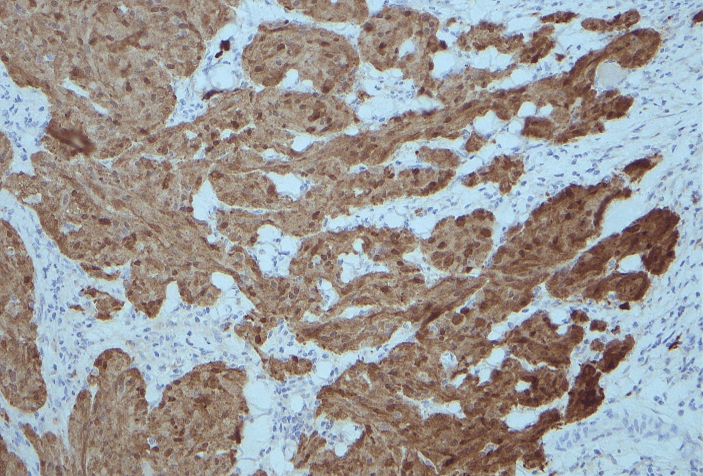
Figure 5: S100 stain showing positive staining of tumour cells.
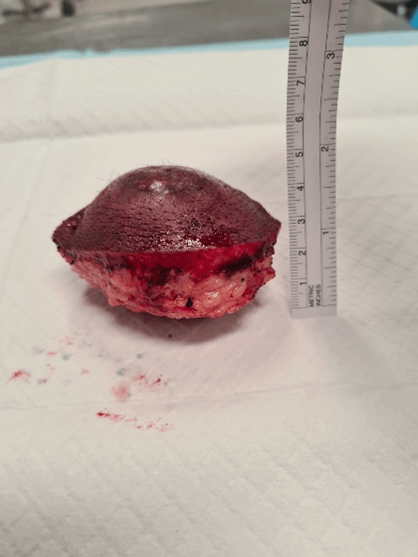
Figure 6: Excised specimen (lateral view).
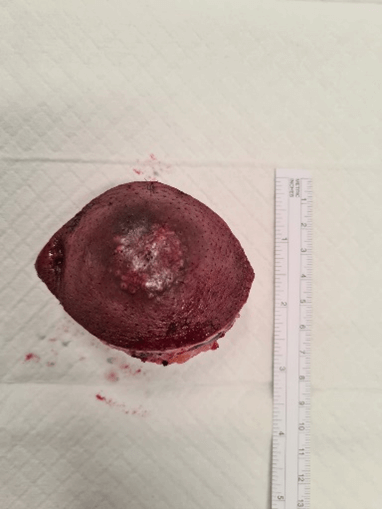
Figure 7: Excised specimen (superior view).
Discussion
Granular cell tumors (previously named Abrikossoff tumors) are rare tumors believed to be derived from Schwann cells [5]. First described in 1926 by the Russian pathologist Abrikossoff and initially thought to be of muscular origin, the introduction of immunohistochemical stains has seen them re-classified as being of Schwann cell origin (positive for S100 and SOX10) [2]. Though rare, S100 negative granular cell tumors have been documented [3].
Granular cell tumors are typically found in the dermis, along mucosal surfaces, and occasionally in skeletal muscle [4]. Clinically they can have the appearance of fibroma, dermatofibrosarcoma protuberans, or basal cell carcinoma [1]. A mean age of 46 years with variable gender predominance is seen, with an increased incidence in darker skin types [1, 6]. They typically present as an asymptomatic slow-growing mass in the head and neck region but can occur anywhere in the body [7]. While usually solitary, multiple lesions are seen in 7% of cases [6]. There is a suggested association with trauma as a history of injury or inflammation is documented in 12.46% of cases [6]. Diagnosis historically occurs after an average of 11 months of symptoms [1]. Size is variable with tumors up to 17cm being described [6]. Imaging characteristics include an echogenic halo, or partially hyperechoic appearance on ultrasound, as well high T2 signal when compared to surrounding muscle, with low to intermediate T1 signal on MRI. However these features are not diagnostic [7].
Multiple benign and malignant lesions may have granular cell features making immunohistochemical stains crucial for accurate diagnosis [4]. Tumors are characterised by infiltrative, non-encapsulated nests, cords, or sheets of polygonal cells, and occasional spindle cells with abundant eosinophilic finely granular cytoplasm [4]. The tumors are characteristically positive for S100, vimentin, NSE, CD56, CD68, SOX10, and inhibin [2, 4]. S100 is a Schwann cell marker and is associated with nearly all peripheral nerve sheath tumors as well as melanomas [8].
Identifying tumors with malignant potential has historically been difficult. When metastasis occurs, outcomes are poor with a median survival of 3 years [4]. Two histological classification systems have been published [4, 9]. The first, developed in 1998 (Fanburg-Smith criteria) divides tumors into benign, atypical, and malignant [4]. The second in 2010 (Nasser-Ahmed-Kowalski), classifies into benign and uncertain malignant potential. This system only designates tumors as malignant when metastasis has been demonstrated [9]. Fanburg-Smith criteria for malignancy include increased nuclear-to-cytoplasmic ratio; nuclear pleomorphism; necrosis; spindling of tumor cells; vesicular nuclei with prominent nucleoli; and mitotic count of more than two in 10 high-power fields, while Nasser-Ahmed-Kowalski consider only the presence of necrosis, and/or mitoses. Both demonstrated a statistical association with higher Ki-67 scores and malignancy, but only Fanburg-Smith et al. showed an association with p53 [4, 9].
A review of 1499 published cases conducted in 2020 suggests a discordance in these classification systems [6]. Based on Fanburg-Smith criteria 71.88% were classified as benign, 16.77% as atypical, and 10.85% as malignant. Based on Nasser-Ahmed-Kowalski classification, 92.77% were benign, and 7.23% had uncertain malignant potential. Evidence of metastatic disease was seen in 2.42% of patients, thus suggesting both systems may over-report malignancy [6]. From the original papers, no recurrence was seen in benign lesions [4, 9]. In malignant disease recommended treatment is excision with wide margins, with mixed evidence for radiation therapy [10].
In this patient, prior to histology, the clinical concern was for dermatofibrosarcoma protuberans due to its firm texture. This uncommon soft tissue tumor also presents as a slow growing, firm plaque or nodule [11]. The experience of misdiagnosis and delayed presentation is not infrequent for granular cell tumors due to their low prevalence and clinical similarity to other soft tissue tumors. They are frequently misdiagnosed as fibromas prior to histological examination [12]. Other case reports include tumors misdiagnosed as epidermoid cysts, as well as furuncles [13, 14]. While exceptionally rare, a diagnosis of granular cell tumor should be considered in the differential diagnosis of an atypical subcutaneous lesion with the above features.
Conflicts of Interest
None.
Funding
None.
Consent
The patient provided written consent for photography as well clinical details to be published for academical and study purposes.
Article Info
Article Type
Case ReportPublication history
Received: Mon 27, Mar 2023Accepted: Wed 19, Apr 2023
Published: Tue 02, May 2023
Copyright
© 2023 Christopher Darlington. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.AJSCR.2023.02.02
Figures & Tables
References
1.
Lack EE, Worsham
GF, Callihan MD, Crawford BE, Klappenbach S et al. (1980) Granular cell tumor: a clinicopathologic
study of 110 patients. J
Surg Oncol 13: 301-316. [Crossref]
2.
An S, Jang J, Min
K, Kim MS, Park H et al. (2015) Granular
cell tumor of the gastrointestinal tract: histologic and immunohistochemical
analysis of 98 cases. Hum
Pathol 46: 813-819. [Crossref]
3.
Cohen JN, Yeh I,
Jordan RC, Wolsky RJ, Horvai AE et al. (2018) Cutaneous Non-Neural Granular Cell Tumors Harbor Recurrent ALK Gene
Fusions. Am J Surg Pathol 42: 1133-1142. [Crossref]
4.
Fanburg Smith JC,
Meis Kindblom JM, Fante R, Kindblom LG (1998) Malignant granular cell tumor of soft tissue: diagnostic criteria and
clinicopathologic correlation.
Am J Surg Pathol 22: 779-794. [Crossref]
5.
Fisher ER, Wechsler
H, Granular cell myoblastoma-a
misnomer. Electron microscopic and histochemical evidence concerning its
Schwann cell derivation and nature (granular cell schwannoma). Cancer 15: 936-954. [Crossref]
6.
Mobarki M,
Dumollard JM, Col PD, Camy F, Peoc’h M et al. (2020) Granular cell tumor a study of 42 cases and systemic review of the
literature. Pathol Res
Pract 216: 152865. [Crossref]
7.
Scaranelo AM,
Bukhanov K, Crystal P, Mulligan AM, O’Malley FP et al. (2007) Granular cell tumour of the breast: MRI
findings and review of the literature.
Br J Radiol 80: 970-974. [Crossref]
8.
Fisher C (2006) The comparative roles of electron microscopy
and immunohistochemistry in the diagnosis of soft tissue tumours. Histopathology 48: 32-41. [Crossref]
9.
Nasser H, Ahmed Y,
Szpunar SM, Kowalski (2011) Malignant
granular cell tumor: a look into the diagnostic criteria. Pathol Res Pract 207: 164-168.
[Crossref]
10.
Mirza FN, Tuggle
CT, Zogg CK, Mirza HN, Narayan D (2018) Epidemiology
of malignant cutaneous granular cell tumors: A US population-based cohort
analysis using the Surveillance, Epidemiology, and End Results (SEER) database. J Am Acad Dermatol 78: 490-497.
[Crossref]
11.
Brooks J, Ramsey ML
(2022) Dermatofibrosarcoma Protuberans.
StatPearls [Internet]. [Crossref]
12.
Torrijos Aguilar A,
Alegre de Miquel V, Pitarch Bort G, Mercader Garcia P, Fortea Baixauli JM
(2009) Cutaneous granular cell tumor:
a clinical and pathologic analysis of 34 cases. Actas Dermosifiliogr 100: 126-132. [Crossref]
13. Choi JH (2020) Smooth Auricular Cutaneous Granular Cell Tumor Mimics Epidermoid Cyst. J Audiol Otol 24: 103-106. [Crossref]
14. Gunduz O, Erkin G, Bilezikçi B, Adanalı G (2016) Slowly Growing Nodule on the Trunk: Cutaneous Granular Cell Tumor. Dermatopathology (Basel) 3: 23-27. [Crossref]