Journals
Primary Site and Other Prognostic Variables Effects on Survival in Pediatric Synovial Sarcoma
A B S T R A C T
Background: Synovial sarcomas (SS) are an aggressive type of soft tissue sarcoma that represent 10% of soft tissue sarcomas. Patients under the age of 20 represent 30% of all SS, and while pediatric and adult patients with SS have similar clinical presentations, pediatric cases have improved outcomes. Prognostic factors include tumor size, primary site, and presence of distant metastases.
Methods: 597 pediatric (<18 years old) patients diagnosed with SS from the National Cancer Database were analyzed. Kaplan-Meier tables were compiled to estimate 1-, 3-, 5-, and 10-year survivals. Groups were compared using log-rank tests and Cox proportional hazards analysis.
Results: Median age at diagnosis was 14 years, 79.4% of patients were Caucasian, and median tumor size was 6.0 cm. The most common anatomical primary site was the extremities, specifically the lower limb and hip. Overall 5- and 10-year survival probabilities were 85.3% and 79.5%, respectively. Tumors in the lungs and thorax had the worst survival, with an overall 5-year survival probability of 50.2%, while upper limb and shoulder tumors had the best 5-year survival probability of 95.9%. The 6-10 age range had the best 5- and 10-year survival probabilities and as age increased the overall survival decreased. Pediatric females had better survival outcomes than males. Approximately 91% of pediatric SS did not present with metastases but the most common site of metastasis was the lungs (2.8%). As histologic grade and tumor stage increased, survival decreases, except that stage II disease showed the best 5-year survival. Biphasic histology had better 5- and 10-year survival outcomes when compared to monophasic histology.
Conclusion: This is the largest and most comprehensive study on pediatric SS to date. Statistically significant prognostic variables of pediatric SS include primary anatomical site, sex, race, histology type, tumor size, and histologic grade and stage.
Keywords
Pediatric Synovial Sarcoma, synovial Sarcoma, prognosis, survivorship, NCDB
Introduction
Soft tissue sarcomas (STS) are tumors derived from mesenchymal origin and represent nearly 8% of all pediatric malignant cancers. Primary soft tissue sarcomas most commonly present in joints, nerves, muscles, fat, deep skin tissues, and blood vessels [1]. Overall the prognosis is better in pediatric soft tissue sarcomas compared to adults [2]. Synovial sarcomas (SS) are an aggressive type of soft tissue sarcoma that represents approximately 10% of soft tissue sarcomas [2]. SS primarily present in the lower extremities, but have been found in many other sites including the abdomen, thorax, and head and neck [3-9]. SS have an estimated incidence rate of 2.75 per 100,000 and are the most common form of non-rhabdomyosarcoma STS [10]. Thirty percent of patients diagnosed with synovial sarcomas are 20 years of age or younger [1].
A study of 150 synovial sarcoma patients under 21 years old found that 5 and 10-year overall survival was 89% and 78%, respectively [11]. Younger age at diagnosis was associated with improved outcomes and the 10-year survival was 90% for patients younger than 20 [11]. Synovial sarcomas have a high incidence of metastasis and the 10-year overall survival of patients with metastatic disease is <50% [11, 12]. The most common site of metastasis for synovial sarcoma is the lung [11]. There are 3 different histopathologic forms of synovial sarcoma divided into two major subtypes: monophasic and biphasic. Monophasic histology is comprised completely of spindle cells, while biphasic includes uniform spindle cells and epithelial cell [13]. Grade and histology have been shown to be a significant prognostic factor for adult synovial sarcoma patients [13, 14]. In a study of SS by Bianchi et al., overall survival was worse with monophasic histology compared to biphasic [13].
One of largest pediatric synovial sarcoma studies to date utilized the Surveillance, Epidemiology and End Results Program (SEER) to compare outcomes of 213 pediatric patients (under 18 years of age) against the adult population. The study looked at primary site, subtype, stage, tumor size, sex and race. The most common anatomical site of origin was the extremities and comparing other sites to the extremities, only adult patients with tumors in the lungs and pleura had a statistically significant increased hazard ratio. The overall 5 and 10-year survival rates for pediatric patients were 83% and 75%, respectively, and as age increased, overall survival probability decreased. Males were found to have worse survival outcomes compared to females [1]. The goal of this study is to compile the largest and most comprehensive cohort of 597 pediatric patients from the National Cancer Database to further characterize the clinicopathologic characteristics and survivorship outcomes in pediatric patients diagnosed with SS between 2004-2015.
Material and Methods
The Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society patterned together in 1989 to create the National Cancer Database (NCDB) [15]. The NCDB is United States oncology database encompassing approximately 70% of all new cancer cases. These cases are added to the database through CoC accredited cancer programs. This data was accessed by the authors through the NCDB’s Participant Use File (PUF) program [15]. The NCDB PUF program was utilized to retrospectively identify 597 patients between the ages of 0 and 18 years diagnosed with single primary synovial sarcoma between 2004 and 2015. The four synovial sarcoma histology subtypes included spindle cell, epithelioid cell, biphasic, and NOS (not otherwise specified) and were identified using ICD-O-3 histology codes 9041, 9042, 9043, and 9040, respectively. Variables extracted from the database included sex, age, race, year of diagnosis, primary anatomical site of tumor, tumor grade, therapies, NCDB analytic stage group and adjuvant therapies. Primary anatomical site of origin was coded using the ICD-O-3 topography codes. The NCDB analytic stage group is comprised of the AJCC pathologic stage. If no pathologic stage was available, the AJCC clinical stage was utilized.
Kaplan Meier methods were used to estimate 1-, 3-, 5-, and 10-year overall survival by primary site, age, sex, grade, NCDB analytic stage, and histology. Log rank tests were used to compare survival curves amongst variables. Patients were grouped together and analyzed by 5 primary sites: head, face and neck; lung and thorax; trunk and pelvis; upper limb and shoulder; and lower limb and hip. Patients were categorized into 4 separate age groups: 0-5 years, 6-10 years, 11-15 years, and 16-18 years. Sex was classified as male or female. Tumor grade was divided into 4 separate groups: well differentiated, differentiated and NOS; moderately differentiated, moderately-well differentiated, and intermediate differentiation; poorly differentiated; and undifferentiated or anaplastic. Stage was analyzed by the NCDB analytic stage group, which compiled the pathologic stages into four separate comprehensive groups: Stage I, Stage II, Stage III, and Stage IV. Histology was broken up into 3 categories: monophasic, biphasic, and synovial sarcoma NOS. Adjuvant therapy was analyzed in 4 categories: surgery alone, surgery with radiation, surgery with chemoradiation, and surgery with systemic therapy. Survival rates were calculated by months between the year of diagnosis until the date of death or last known date of contact. Kaplan-Meier survival curves and descriptive statistics were created using SPSS version 25. Cox proportional hazard regression analysis was performed in SPSS version 25 to calculate hazard ratios and 95% confidence intervals.
Table 1: Epidemiologic Variables of 597 Pediatric Patients with Synovial Sarcoma
|
Variable |
N |
% of total |
|
Age (years) |
|
|
|
0-5 |
18 |
3.0 |
|
6-10 |
86 |
14.4 |
|
11-15 |
269 |
45.1 |
|
16-18 |
224 |
37.5 |
|
Year of Diagnosis |
|
|
|
2004-2009 |
315 |
52.8 |
|
2010-2015 |
282 |
47.2 |
|
Sex |
|
|
|
Male |
319 |
53.4 |
|
Female |
278 |
46.6 |
|
Race |
|
|
|
Caucasian |
474 |
79.4 |
|
African American |
56 |
9.4 |
|
Asian |
19 |
3.2 |
|
Asian Indian, or Pakistani |
6 |
1 |
|
American Indian, Aleutian or Eskimo |
3 |
0.5 |
Results
Clinical characteristics are displayed in (Table 1). The median age at diagnosis was 14 years old. There were 315 cases of pediatric synovial sarcoma diagnosed in the time period of 2004-2009 and 282 cases diagnosed from 2010-2015. The ratio of male to female was 1.12 to 1 with 319 male and 278 female patients. The largest race represented was Caucasian patients comprising 79.4% (n = 474) of the patient population, while African American patients accounted for the second highest population with 56 patients (9.4%) of the patient population. Asian patients consisted of the third largest group with 19 patients (3.2%) and included Chinese, Japanese, Filipino, Vietnamese, and Laotian patients.
Tumor characteristics are shown in (Table 2). Synovial Sarcoma NOS was the most common histology (38.7%), followed by monophasic (33.5%). The most common anatomical primary sites were the lower limb and hip (46.7%), followed by the upper limb and shoulder (21.8%). The median tumor size was 6.0 cm. The majority (75%) of tumors were less than 10 cm. Approximately 27.6% of pediatric synovial sarcomas tumors were poorly-differentiated and 18.6% were moderately-differentiated. Stage II was the most common presentation at 23.1%, followed closely by Stage I (22.3%). Table 3 displays the metastatic characteristics and therapy options for synovial sarcoma patients. The majority of cases were not metastatic (90.5%). No patients had distant lymph node involvement but 6.4% of patients had distant metastases to other organs. The most common site of metastasis was lung involvement, affecting 2.8% of patients. Lymphatic or vascular invasion represented 1.8% of metastases and there were only 2 patients with bone metastases (0.3%). There were no brain or liver metastases.
Table 2: Tumor Characteristics of 597 Pediatric Patients with Synovial Sarcoma
|
Variable |
N |
% of Total |
|
Histology |
|
|
|
Monophasic Spindle Cell |
200 |
33.5 |
|
Epithelioid Cell |
1 |
0.2 |
|
Biphasic |
165 |
27.6 |
|
Synovial Sarcoma NOS** |
231 |
38.7 |
|
Primary Site of Cancer* |
|
|
|
Head, Face, and Neck |
51 |
8.5 |
|
Lung |
7 |
1.2 |
|
Heart and Mediastinum |
7 |
1.2 |
|
Upper Limb and Shoulder |
130 |
21.8 |
|
Lower Limb and Hip |
279 |
46.7 |
|
Thorax |
24 |
4.0 |
|
Abdomen |
29 |
4.9 |
|
Pelvis |
29 |
4.9 |
|
Trunk, NOS** |
17 |
2.8 |
|
Overlapping lesion |
7 |
1.2 |
|
Other soft tissues, NOS** |
11 |
1.8 |
|
Peripheral nerves and autonomic nervous system of head, face and neck |
1 |
0.2 |
|
Peripheral nerves and autonomic nervous system of pelvis, lower limb, and hip |
5 |
0.8 |
|
Tumor size |
|
|
|
< 5 cm |
250 |
41.9 |
|
5-10 cm |
199 |
33.3 |
|
> 10 cm |
76 |
12.7 |
|
Unknown |
72 |
12.1 |
|
Tumor grade |
|
|
|
Well-differentiated |
15 |
2.5 |
|
Moderately-differentiated |
111 |
18.6 |
|
Poorly-differentiated |
165 |
27.6 |
|
Undifferentiated, anaplastic |
48 |
8.0 |
|
Cell type not determined |
258 |
43.2 |
|
NCDB Analytical Stage |
|
|
|
Stage I |
133 |
22.3 |
|
Stage II |
138 |
23.1 |
|
Stage III |
101 |
16.9 |
|
Stage IV |
34 |
5.7 |
|
Staging unknown or not applicable |
191 |
32.0 |
NOS: Not otherwise specified
*Anatomical sites include connective, subcutaneous and other soft tissue of:
Surgery was the most common treatment modality with 91.1% of patients electing to have surgery, while 58.6% of patients were treated with radiation and 54.4% underwent chemotherapy. Approximately 56.8% of patients had surgery alone, 24.6% had radiation after surgery, 11.8% of patients had chemoradiation after surgery, and 6.8% had systemic therapy after surgery. Table 4 displays the 1-, 3-, 5-, and 10-year survival probabilities and overall survival for primary site, age, sex, grade, stage, histology, tumor size, and adjuvant therapy. These are graphically displayed in (Figures 1-9). As shown in (Figure 1), the median overall survival was 60.4 months with 1-,3-,5-, and 10-year survival probabilities of 98.1%, 89.3%, 85.3%, and 79.5%, respectively.
Figure 2 displays survival probabilities by primary site. Upper limb and shoulder primary site had the best 1-,3-,5-, and 10-year survival probabilities of 100%, 98.2%, 95.9%, and 86.7%, respectively, followed in decreasing order by lower limb and hip; head, face, and neck; trunk and pelvis; and finally lung and thorax with the poorest 1-,3-,5-, and 10-year survival probabilities of 89.7%, 59.8%, 50.2%, and 50.2 %, respectively. Figure 3 shows overall survival by age and the 6-10 year age group had the best 1-,3-, 5-, and 10-year survival probabilities with 100%, 98.6%, 98.6%, and 96%, respectively. The worst 5-year survival probabilities were found in the age groups 16-18 years while the worst 10 year survival was seen in the 0-5 years group. Figure 4 shows that females had better outcomes than males with 1-,3-,5-, and 10-year survival probabilities of 98.8%, 93.2%, 90.3% and 85.4% compared to 97.5%, 85.7%, 80.7%, and 74.2%, respectively.
Table 3: Metastasis and Therapy Characteristics for Pediatric Synovial Sarcoma Patients
|
Variable |
N= 597 |
% of Total |
|
Metastases Present at Diagnosis |
|
|
|
No distant metastasis |
540 |
90.5 |
|
Distant lymph node(s) |
0 |
0 |
|
Distant metastasis except distant lymph node(s), Carcinomatosis |
31 |
5.2 |
|
Distant metastasis plus distant lymph nodes |
7 |
1.2 |
|
Unknown |
19 |
3.2 |
|
Location of Metastases |
|
|
|
Bone |
2 |
0.3 |
|
Lung |
17 |
2.8 |
|
Liver |
0 |
0 |
|
Brain |
0 |
0 |
|
Lymphatic or Vascular Invasion |
11 |
1.8 |
|
Treatment modality |
|
|
|
Surgery |
544 |
91.1 |
|
Radiation |
350 |
58.6 |
|
Chemotherapy |
325 |
54.4 |
|
Adjuvant Therapy |
N=544 |
% of Total |
|
Surgery Only |
309 |
56.8 |
|
Surgery with Systemic Therapy |
37 |
6.8 |
|
Surgery with Radiation |
134 |
24.6 |
|
Surgery with Chemoradiation |
64 |
11.8 |
Table 4: 1-, 3-, 5- and 10-Year Survival Probabilities for Primary Site, Age, Sex, Grade, Stage, Histology, Tumor Size, and Adjuvant Therapy
|
Variable |
Probability of 1-Year Survival (%) |
Probability of 3-Year Survival (%) |
Probability of 5-Year Survival (%) |
Probability of 10-Year Survival (%) |
|
Overall Survival |
98.1 |
89.3 |
85.3 |
79.5 |
|
Primary Anatomical Site |
|
|
|
|
|
Head, Face and Neck |
100 |
88.0 |
84.5 |
79.8 |
|
Lung and Thorax |
89.7 |
59.8 |
50.2 |
50.2 |
|
Trunk and Pelvis |
98.6 |
87.2 |
81.2 |
71.1 |
|
Upper Limb and Shoulder |
100 |
98.2 |
95.9 |
86.7 |
|
Lower Limb and Hip |
98.4 |
89.0 |
85.0 |
80.8 |
|
Age Group (years) |
|
|
|
|
|
0-5 |
93.8 |
93.8 |
93.8 |
62.5 |
|
6-10 |
100 |
98.6 |
98.6 |
96.0 |
|
11-15 |
99.2 |
91.4 |
87.7 |
82.3 |
|
16-18 |
95.9 |
82.8 |
76.8 |
71.6 |
|
Sex |
|
|
|
|
|
Male |
97.5 |
85.7 |
80.7 |
74.2 |
|
Female |
98.8 |
93.2 |
90.3 |
85.4 |
|
Grade |
|
|
|
|
|
Well Differentiated, Differentiated |
100 |
100 |
100 |
100 |
|
Moderately differentiated, moderately well differentiated, intermediate differentiation |
100 |
96.6 |
95 |
90.5 |
|
Poorly differentiated |
97.2 |
83.6 |
78.9 |
76.0 |
|
Undifferentiated, anaplastic |
95.3 |
77.2 |
70.6 |
61.7 |
|
NCDB Analytical Stage |
|
|
|
|
|
Stage I |
100 |
95.2 |
91.7 |
90.1 |
|
Stage II |
100 |
98.1 |
93.7 |
79.2 |
|
Stage III |
97.8 |
85.6 |
82.2 |
72.2 |
|
Stage IV |
87.0 |
31.3 |
27.4 |
27.4 |
|
Histology |
|
|
|
|
|
Synovial Sarcoma, NOS |
96.0 |
81.6 |
76.7 |
67.1 |
|
Monophasic Synovial Sarcoma |
98.9 |
90.6 |
87.7 |
82.1 |
|
Biphasic Synovial Sarcoma |
100 |
97.8 |
93.8 |
93.8 |
|
Tumor Size |
|
|
|
|
|
<5 cm |
100 |
96.5 |
95.7 |
92.6 |
|
5-10 cm |
99.4 |
90.5 |
82.6 |
73.5 |
|
>10 cm |
94.7 |
75.9 |
73.4 |
73.4 |
|
Adjuvant Therapy |
|
|
|
|
|
Surgery Only |
98.9 |
92.0 |
89.4 |
86.2 |
|
Surgery with Systemic Therapy |
100 |
93.5 |
79.7 |
79.7 |
|
Surgery with Radiation |
99.2 |
91.4 |
88.3 |
81.3 |
|
Surgery with Chemoradiation |
98.1 |
86.4 |
81.3 |
76.6 |
*NOS: Not Otherwise Specified
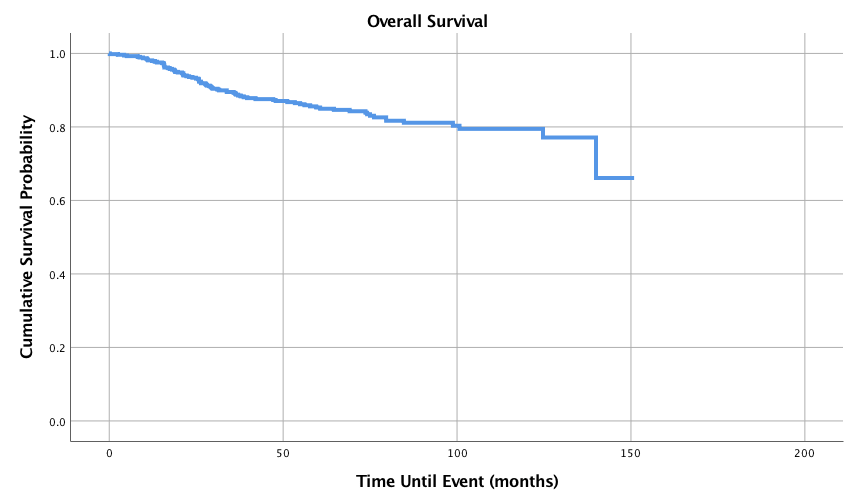
Figure 1: Overall Survival for Pediatric Patients with Synovial Sarcoma, n = 545
Graphed survival probabilities by grade and stage are shown in (Figures 5 and 6) respectively. Well differentiated grades of tumors had a 1-,3-,5-, and 10-year survival probability of 100%, while undifferentiated or anaplastic grades had the worst 1-,3-, 5-, and 10-year survival probabilities of 95.3%, 77.2%, 70.6% and 61.7%, respectively. Stage II tumors had the best 5-year survival probability (93.7%) and stage I had the best 10-year survival probability (90.1%). Stage IV had the worst 5 and 10-year survival probabilities with 27.4% as both the 5 and 10-year survival probability. Kaplan-Meier curves of the different histology types is shown in (Figure 7). Biphasic synovial sarcoma had the best 1-,3-, 5-, and 10-year survival probabilities at 100%, 97.8%, 93.8%, and 93.8%, respectively. Synovial Sarcoma NOS had the lowest 5 and 10-year survival probabilities of 76.7% and 67.1%, respectively.
As shown in (Figure 8), smaller tumor size (<5cm) was associated with improved 1-,3-,5-, and 10-year survival outcomes of 100%, 96.5%, 95.7% and 92.6%. Tumors measuring 5-10 cm had 3-, 5-, and 10-year survival rates of 90.5%, 82.1% and 73%, respectively and >10 cm had the worst 3-, 5-, and 10-year survival probabilities (75.9%, 73.4% and 73.4%, respectively). Figure 9 shows survival curves based on different adjuvant treatment modalities. Out of the adjuvant therapies, surgery alone had the best 5- and 10-year survival probabilities of 89.4 and 86.2% respectively, while surgery with radiation had the second-best overall survival (5-year 88.3% and 10-year 81.3%). Surgery with systemic therapy had the worst 5-year rate (79.7%), however had the best 1- and 3-year survival probabilities of 100% and 93.5%, respectively. Surgery with chemoradiation had the worst 10-year overall survival (76.6%). Table 5 displays hazard ratios for all of the studied variables.
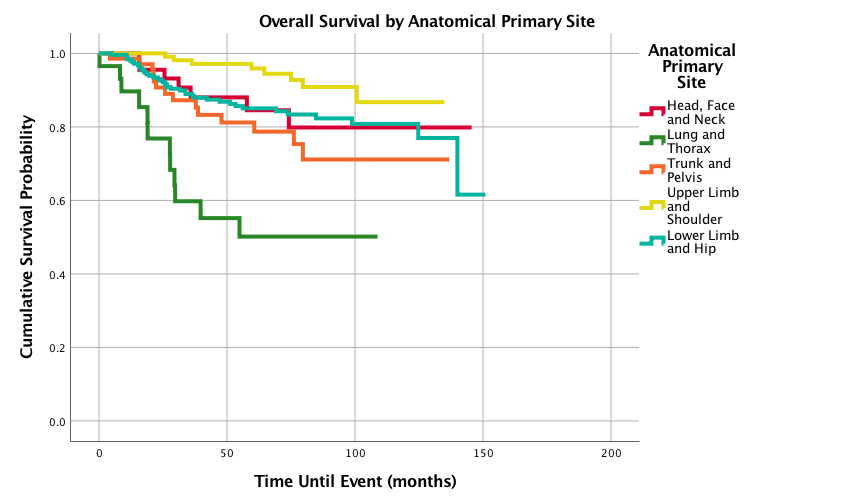
Figure 2: Overall Survival for Pediatric Patients with Synovial Sarcoma by Anatomical Primary Site, n = 528, p<0.0005
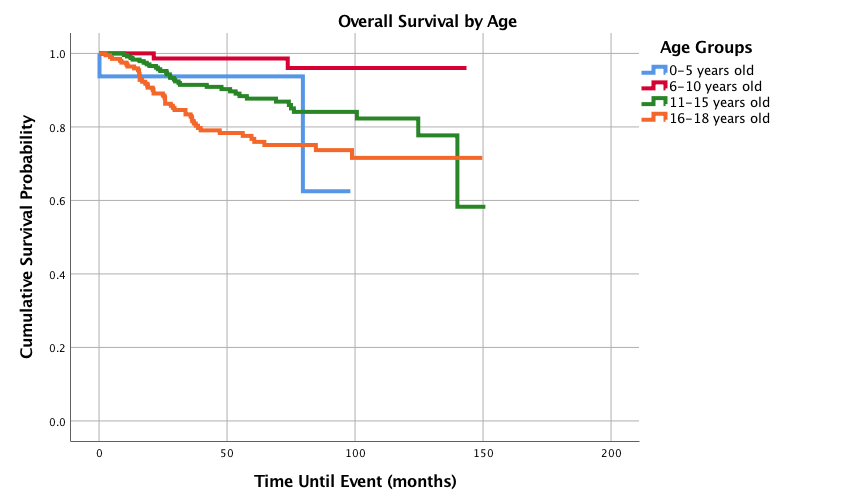
Figure 3: Overall Survival for Pediatric Patients with Synovial Sarcoma by Age, n = 545, p<0.0005
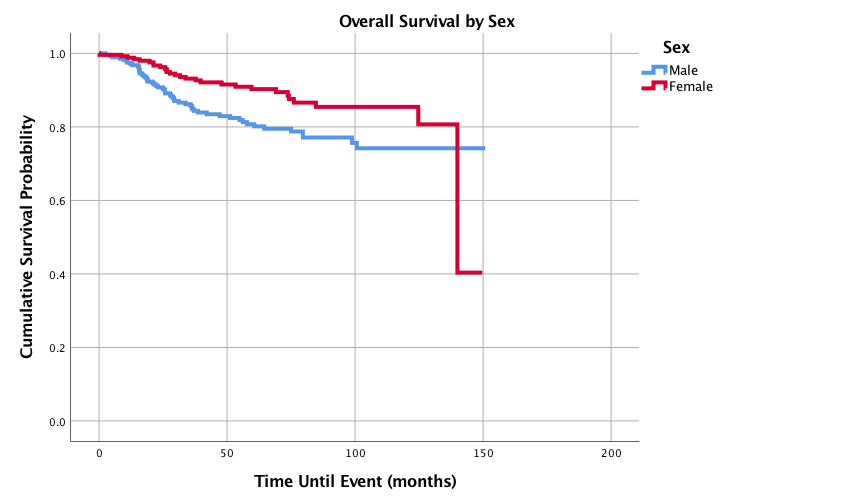
Figure 4: Overall Survival for Pediatric Patients with Synovial Sarcoma by Sex, n = 545, p=0.009
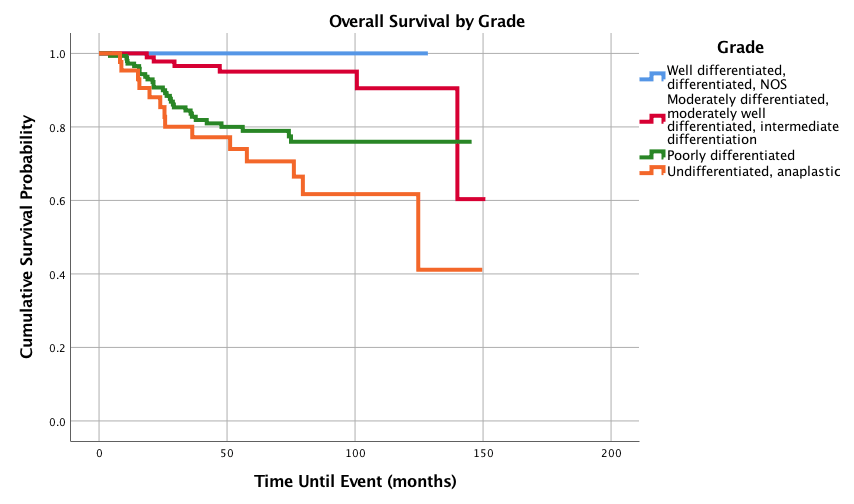
Figure 5: Overall Survival for Pediatric Patients with Synovial Sarcoma by Grade, n = 306, p<0.0005
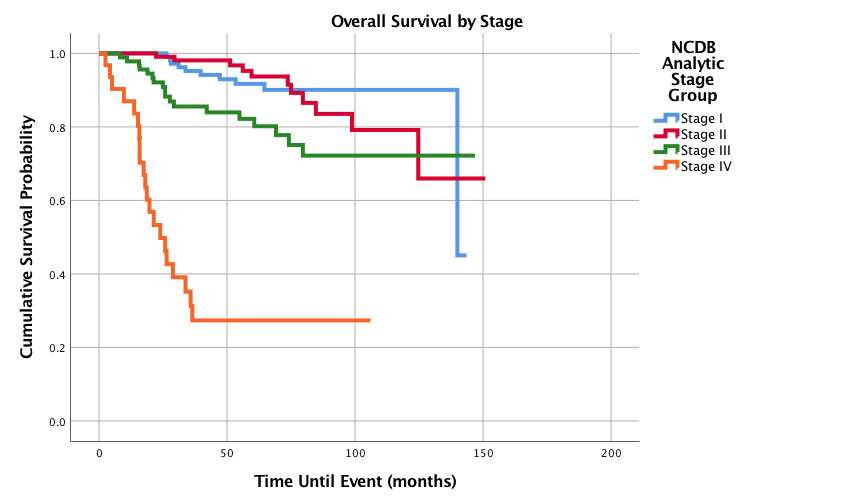
Figure 6: Overall Survival for Pediatric Patients with Synovial Sarcoma by Stage, n = 365, p<0.0005
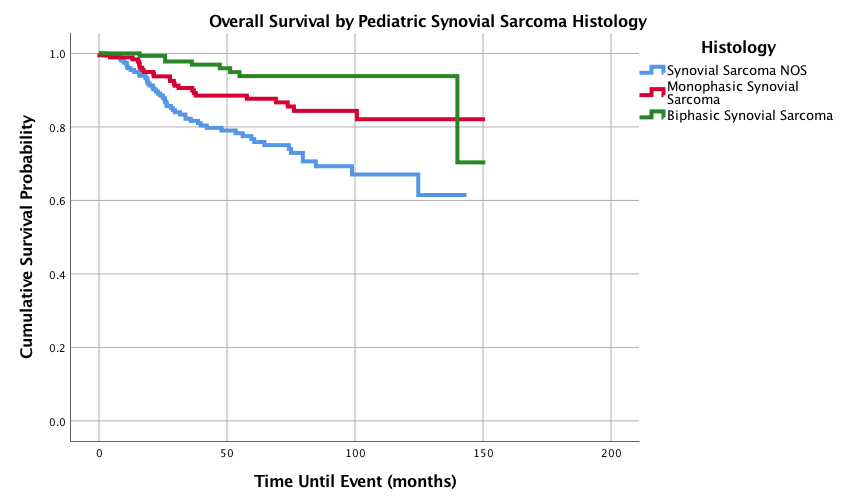
Figure 7: Overall Survival for Pediatric Patients with Synovial Sarcoma by histology, n = 545, p<0.0005.
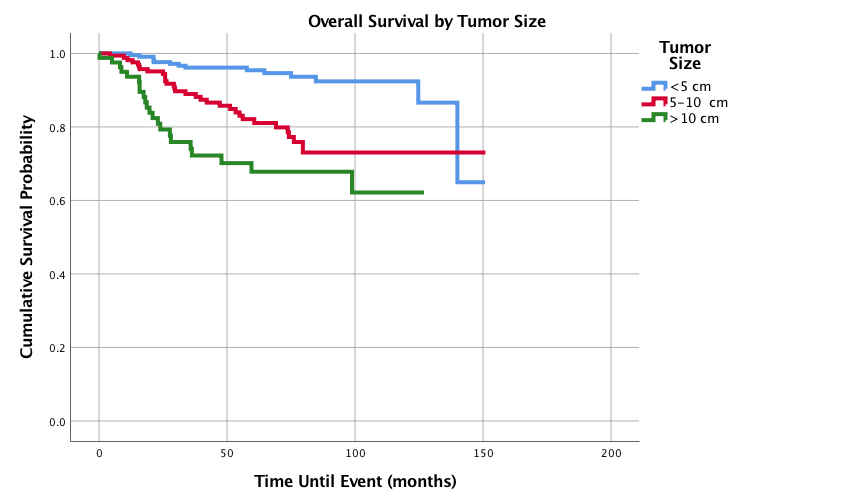
Figure 8: Overall Survival for Pediatric Patients with Synovial Sarcoma by Tumor size, n = 479, p<0.0005.
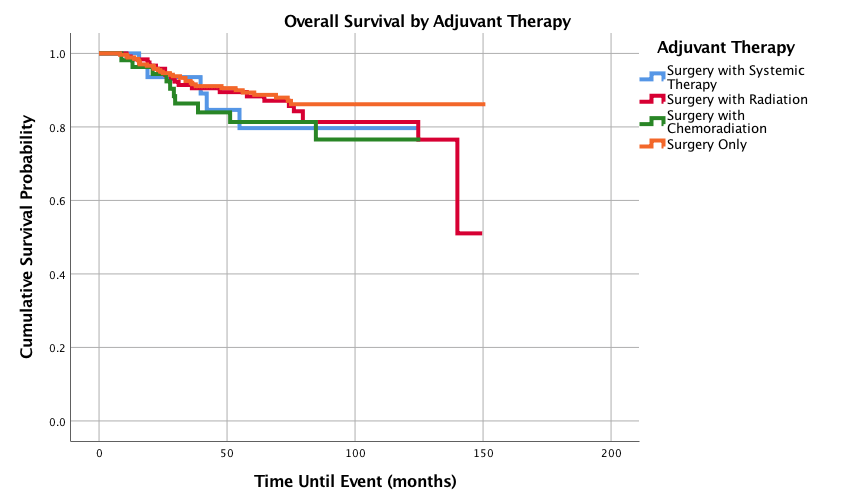
Figure 9: OOverall Survival for Pediatric Patients with SS by Adjuvant Therapy, n = 494, p=0.385.
Table 5: Hazard Ratios for Primary Site, Age, Sex, Race, Histology, Tumor Size, Grade, Stage, and Adjuvant Therapy
|
Variable |
Hazard Ratio (95% Confidence Interval) |
P-value |
|
Primary Anatomical Site |
|
|
|
Upper Limb and Shoulder |
Reference |
|
|
Head, Face and Neck |
2.374 (0.858-6.568) |
0.096 |
|
Lung and Thorax |
10.021 (4.089-24.563) |
<0.0005 |
|
Trunk and Pelvis |
3.625 (1.520-8.646) |
0.004 |
|
Lower Limb and Hip |
2.500 (1.167-5.355) |
0.018 |
|
Age Category |
|
|
|
6-10 years |
Reference |
|
|
11-15 years |
2.290 (0.509-10.308) |
0.280 |
|
16-18 years |
4.191 (0.946-18.560) |
0.059 |
|
Sex |
|
|
|
Female |
Reference |
|
|
Male |
2.251 (1.392-3.640) |
0.001 |
|
Race |
|
|
|
Caucasian |
Reference |
|
|
African American |
1.526 (.804-2.898) |
0.196 |
|
Asian |
2.386 (1.026-5.545) |
0.043 |
|
Histology |
|
|
|
Biphasic |
Reference |
|
|
Monophasic Spindle Cell |
0.473(0.283-0.789) |
0.022 |
|
Synovial Sarcoma NOS |
5.276 (2.496-11.152) |
<0.0005 |
|
Tumor size |
|
|
|
< 5 cm |
Reference |
|
|
5-10 cm |
3.473 (1.868-6.457) |
<0.0005 |
|
> 10 cm |
6.323 (3.163-12.642) |
<0.0005 |
|
Tumor grade |
|
|
|
Moderately-differentiated |
Reference |
|
|
Poorly-differentiated |
2.082 (0.832-5.209) |
0.117 |
|
Undifferentiated, anaplastic |
3.441 (1.283-9.229) |
0.014 |
|
NCDB Analytical Stage |
|
|
|
Stage I |
Reference |
|
|
Stage II |
1.091 (0.449-2.651) |
0.847 |
|
Stage III |
2.313 (1.004-5.331) |
0.049 |
|
Stage IV |
15.062 (6.526-34.761) |
<0.0005 |
|
Adjuvant Therapy |
|
|
|
Surgery Only |
Reference |
|
|
Surgery with Systemic Therapy |
1.539 (.595-3.982) |
0.374 |
|
Surgery with Radiation |
1.345 (.759-2.381) |
0.310 |
|
Surgery with Chemoradiation |
1.775 (0.864-3.647) |
0.118 |
Discussion
This is the largest and most comprehensive study to evaluate pediatric patients with synovial sarcoma. Synovial sarcoma is a rare tumor, with 30% of cases thought to occur in patients under 20 [16]. This study demonstrated a potentially lower prevalence, as synovial sarcomas represent 0.05% of NCDB patients from 2004-2015, with 11.8% of those patients being pediatric patients. In this study cohort, pediatric SS had a median age of 14, affected males (53%) and females (47%) nearly equally, and was most common in Caucasian (79.4%) followed by African American (9.4%) patients. These findings are largely supported by previous studies [12, 17, 18]. This current study had a slightly higher percentage of Caucasian patients and a lower percentage of African American patients when comparing our study population to the US population. (Caucasian-76.9%, African American-13.3%; United States Census) [19].
The most common histology was synovial sarcoma NOS with 38.7%, while monophasic spindle cell was next at 33.5%. This may represent different pathology reporting practices and after disregarding NOS, most of the previous studies show monophasic histology comprised the largest histologic group [1, 13]. This study reaffirmed previous findings in the literature with the highest percentage of primary pediatric synovial sarcomas located in the extremities, specifically the lower leg and hip [1, 16]. These tumors presented early with Stage II disease most common, followed closely by stage I. These findings were also noted by Sultan [1]. The most common grade that presented was poorly differentiated. Bianchi used a different grading system but found that grade 3 was most common and had the worst survival [13].
The majority (90.5%) of patients presented without metastasis. The most common site of metastasis was the lung (2.8% of all patients but accounting for 44.7% out of patients with distant metastasis), consistent with existing literature [11, 16]. The majority (91.1%) of patients received surgery as part of their treatment, followed by radiation (58.6%), and chemotherapy (54.4%). This is similar to prior epidemiologic studies in both pediatric and adult populations [1, 13].
Age has been well documented as an important prognostic indicator in both adult and pediatric SS. This study demonstrated a 5- and 10- year survival of 85.3% and 79.5%, respectively in an exclusive pediatric population. This is similar to past studies in pediatric populations, in which five-year overall survival probabilities have been estimated between 80-83% and ten-year pediatric SS OS estimates of 77% [1, 16, 20]. To the best of our knowledge, there has been no previous study that has broken down pediatric patients into different age groups to look at 1-,3-, 5- or 10-year survival probability. It has been well studied that overall survival decreases with increasing age [1, 16, 20]. Sultan et al. also found that adolescents had worse outcomes compared to children less than 10 years of age, however no 5- and 10-year survival probabilities were provided [1]. This current study found that the 6-10 year old cohort had improved 5-year overall survival, followed by 0-5 years old, 11-15 years and finally the 16-18 years. Interestingly, the 6-10 year old cohort still maintained the most favorable 10-year overall survival, while the 0-5 year old age group dropped down to become the least favorable outcome of the four groups. When compared to the 6-10 year old patients, age groups of 11-15 and 16-18 had increased hazard ratios, however neither rose to statistical significance, while the 0-5 age group was excluded from analysis due to small sample size. Small sample size may also play a role in the marked drop from 5- to 10-year survival in the 0-5 year old cohort, but may warrant further study. It has been noted that patients under the age of 10 are more likely to present with favorable clinical features, notably tumor size under 5cm, primary site of the extremity, and localized presentation [1]. More research to distinguish the specific clinicopathologic characteristics in the 6-10 year old cohort may help identify favorable traits to better target therapies for all age groups.
Previous studies have shown that African American pediatric patients with SS had worse survival outcomes compared to Caucasian patients [1]. In our study, both African American and Asian patients had increased hazard ratios compared to Caucasian patients, but only Asian patients reached statistical significance. Sultan et al. found that female patients with SS had better survival rates among adults but was not seen in pediatric patients [1]. This current study demonstrated through log rank analysis that female pediatric patients had significantly better 1-,3- 5-, and 10-year survival probabilities compared to their male counterparts and males had a statistically significant increased hazard ratio when compared to females.
This is the most comprehensive study on primary anatomical sites in pediatric SS patients. A previous study found a difference in survival between extremity and axial SS pediatric patients, however it only presented 5-year survival probabilities on 115 patients with 30 patients grouped into an axial primary site category [12]. Further survival analysis was necessary to characterize 1-,3-,5-, and 10-year survival probabilities by primary anatomical site. Ferrari found a 5-year survival probability for pediatric SS extremity tumors to be 84%, while our current study found a similar 5-year survival for patients with lower limb and hip tumors at 85% and a much higher 5-year survival of 95.9% for the upper limb and shoulder anatomical site [12]. Pediatric synovial sarcoma tumors in the upper limb and shoulder had the best survival outcomes followed in decreasing order by lower limb and hip; head, face, and neck; trunk and pelvis; and finally, lung and thorax. When comparing other primary sites to the upper limb and shoulder, all the other primary sites had increased hazard ratios. Statistically significant increased hazard ratios were noted at the other primary sites in increasing order: lower limb and hip (2.5); trunk and pelvis (3.625); and most notably lung and thorax (10.021).
The effect of tumor grade on prognosis has not been studied in pediatric SS populations, but grade has been found to have an effect on survival probabilities in a previous study that consisted of 177 adults and 19 pediatric patients [13]. It is difficult to compare, as Bianchi’s study utilized the French Federation of Cancer Centers Sarcoma Group (FNLCC) grading system based on tumor differentiation, mitotic count and necrosis, while the NCDB utilizes a four-tiered grading system based on differentiation status. However, Bianchi et al. found that increasing grade resulted in lower survival probabilities and the highest grade (grade III) had a 10-year survival of 42.8% [13]. Our current study found that among pediatric patients, as grade increases probabilities of 1-,3-, 5-, and 10-year survival significantly decreases and the 10-year survival for the highest grade (undifferentiated or anaplastic) dropped to 61.7%. Compared to moderately differentiated grades, while both poorly differentiated and undifferentiated/anaplastic had increased hazard ratios, only undifferentiated/anaplastic rose to statistical significance.
Not unexpectedly, this study found that as stage at initial diagnosis increases, the overall 5- and 10-year survival probabilities decrease. Somewhat surprisingly, Stage II had the best 3- and 5-year survival rates and Stage I had the best 10-year survival rate. For stage IV, only 1 in every 4 patients survives at both 5 and 10 years. Comparing the other stages to stage I, Stage III and Stage IV had a statistically significant increased hazard ratio, with Stage IV showing an impressive ratio of 15.1.
Histologic type has been determined as a significant prognostic variable with both monophasic and SS NOS histology showing increased hazard ratios when compared to biphasic histology. This study suggests improved 5- and 10- year survivals with biphasic (93.8%, 93.8%) compared to monophasic (87.7%, 82.1%) histology. The improved biphasic histology survival was also noted by Okcu et al, in a study of 219 SS patients under the age of 20 (biphasic 78%, monophasic 67%), although the survival probabilities reported here are more encouraging. Ten-year survival based on histology has not previously discussed in the literature [16].
As tumor size increased, both the overall 5- and 10-year survival decreased. This has also been found in pediatric patients in the SEER analysis, however there were only two groups in that previous study: <5 cm and >5 cm [1]. This current study formed 3 groups for tumor size (<5 cm, 5-10 cm, and >10 cm) to better characterize tumor size as a prognostic factor in pediatric patients. Tumors larger than 10cm had the worst 5- and 10-year overall survival. Tumors sized between 5-10 cm and those larger than 10 cm both had statistically significant increased hazard ratios, with tumor sizes >10 cm having almost double the hazard ratio of tumors sized between 5-10 cm.
Adjuvant therapies are more frequently used in pediatric than adult populations [12]. Site surgery alone was the most employed therapy strategy. In adjuvant settings, radiation was the most common adjuvant therapy (24.6%) and systemic therapy was the least chosen with 6.8% in a study by Ferrari et al in 2004. In our current study, adjuvant radiotherapy had the best 5- and 10-year survival probabilities of 88.3% and 81.3%, respectively. This survivorship is higher in our pediatric cohort compared to overall estimates for all synovial sarcoma patients. A study by Song et al identified 103 synovial sarcoma patients with a median age of 33 years old. Those patients were older and may have had more aggressive lesions, but 73% of patients received adjuvant radiotherapy, and 5- and 10- year overall survival was 77 and 65%, respectively (Song et al.). The role of adjuvant chemotherapy remains under investigation. A 2017 NCDB study by Vining et al determined that adjuvant chemotherapy was associated with improved overall survival in Stage III patients, but not in Stage I or Stage II [21]. Future studies on the pediatric synovial sarcoma cohort may better characterize the subset of patients receiving each treatment strategy and help optimize the overall use of these therapies.
Current recommendations on treatment of pediatric synovial sarcoma is based on risk stratification, determined by tumor size, primary site, and clinical group. Tumor size greater than 5cm and primary site on the head and neck, trunk or pleura are considered high risk. Surgical resection alone is recommended for low-risk groups, and chemotherapy is recommended for high risk groups. Overall survival for chemotherapy administered to high risk groups was 74% [2, 12]. Although our current study does not assess cases by risk, our findings help support the rationale for this approach. Further studies may utilize the NCDB to analyze adherence to these recommendations, and the guidelines’ efficacy. This work has several limitations inherent in retrospective, large database analyses. While distinct advantages of this NCDB study include a very comprehensive dataset, high standard of inclusion, and decreased likelihood of selection bias. Another major disadvantage is potential reporting error. Multiple independent registrars collect and report the data. Data can be incompletely, improperly, or inaccurately reported (Participant User Files). Survivorship data is also limited to overall survival, so deaths from unrelated causes are included.
In conclusion, this is the largest and most comprehensive study on pediatric synovial sarcoma to date. Significant clinical prognostic variables of pediatric synovial sarcoma include primary anatomical site, sex, race, tumor size, histology, tumor grade, and tumor stage. Negative prognostic factors include primary tumors in the lungs and thorax, male sex, Asian race, monophasic spindle cell and biphasic histology, undifferentiated or anaplastic tumor grades, and stage III and IV tumors. Primary site surgery alone was the most frequently employed treatment strategy. Adjuvant radiotherapy was associated with the best survival outcomes compared to adjuvant chemoradiation or adjuvant systemic therapy.
Funding
This manuscript had no funding sources
Conflicts of interest
There is not conflict of interest.
Author Contributions
Conceptualization, J.G.; Methodology, J.G., C.C. and P.S..; Investigation, J.G.; Formal Analysis and Visualization, J.G.; Writing-Original Draft J.G. and I.G.; Writing-Review and Editing, C.C and P.S.
Significance
This study utilized the NCDB to compile the largest and most comprehensive cohort of pediatric (<18 years old) patients with synovial sarcoma to retrospectively summarize significant prognostic variables and their effects on survival to improve clinical outcomes worldwide.
Discipline
Soft Tissue and Bone Sarcoma
Article Info
Article Type
Research ArticlePublication history
Received: Thu 16, May 2019Accepted: Fri 12, Jul 2019
Published: Thu 08, Aug 2019
Copyright
© 2023 Peter Silberstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JSO.2019.02.10
Author Info
Corresponding Author
Peter SilbersteinDepartment of Internal Medicine, Division of Hematology/Oncology, Creighton University Medical Center, Omaha, NE, USA
Figures & Tables
Table 1: Epidemiologic Variables of 597 Pediatric Patients with Synovial Sarcoma
|
Variable |
N |
% of total |
|
Age (years) |
|
|
|
0-5 |
18 |
3.0 |
|
6-10 |
86 |
14.4 |
|
11-15 |
269 |
45.1 |
|
16-18 |
224 |
37.5 |
|
Year of Diagnosis |
|
|
|
2004-2009 |
315 |
52.8 |
|
2010-2015 |
282 |
47.2 |
|
Sex |
|
|
|
Male |
319 |
53.4 |
|
Female |
278 |
46.6 |
|
Race |
|
|
|
Caucasian |
474 |
79.4 |
|
African American |
56 |
9.4 |
|
Asian |
19 |
3.2 |
|
Asian Indian, or Pakistani |
6 |
1 |
|
American Indian, Aleutian or Eskimo |
3 |
0.5 |
Table 2: Tumor Characteristics of 597 Pediatric Patients with Synovial Sarcoma
|
Variable |
N |
% of Total |
|
Histology |
|
|
|
Monophasic Spindle Cell |
200 |
33.5 |
|
Epithelioid Cell |
1 |
0.2 |
|
Biphasic |
165 |
27.6 |
|
Synovial Sarcoma NOS** |
231 |
38.7 |
|
Primary Site of Cancer* |
|
|
|
Head, Face, and Neck |
51 |
8.5 |
|
Lung |
7 |
1.2 |
|
Heart and Mediastinum |
7 |
1.2 |
|
Upper Limb and Shoulder |
130 |
21.8 |
|
Lower Limb and Hip |
279 |
46.7 |
|
Thorax |
24 |
4.0 |
|
Abdomen |
29 |
4.9 |
|
Pelvis |
29 |
4.9 |
|
Trunk, NOS** |
17 |
2.8 |
|
Overlapping lesion |
7 |
1.2 |
|
Other soft tissues, NOS** |
11 |
1.8 |
|
Peripheral nerves and autonomic nervous system of head, face and neck |
1 |
0.2 |
|
Peripheral nerves and autonomic nervous system of pelvis, lower limb, and hip |
5 |
0.8 |
|
Tumor size |
|
|
|
< 5 cm |
250 |
41.9 |
|
5-10 cm |
199 |
33.3 |
|
> 10 cm |
76 |
12.7 |
|
Unknown |
72 |
12.1 |
|
Tumor grade |
|
|
|
Well-differentiated |
15 |
2.5 |
|
Moderately-differentiated |
111 |
18.6 |
|
Poorly-differentiated |
165 |
27.6 |
|
Undifferentiated, anaplastic |
48 |
8.0 |
|
Cell type not determined |
258 |
43.2 |
|
NCDB Analytical Stage |
|
|
|
Stage I |
133 |
22.3 |
|
Stage II |
138 |
23.1 |
|
Stage III |
101 |
16.9 |
|
Stage IV |
34 |
5.7 |
|
Staging unknown or not applicable |
191 |
32.0 |
NOS: Not otherwise specified
*Anatomical sites include connective, subcutaneous and other soft tissue of:
Table 3: Metastasis and Therapy Characteristics for Pediatric Synovial Sarcoma Patients
|
Variable |
N= 597 |
% of Total |
|
Metastases Present at Diagnosis |
|
|
|
No distant metastasis |
540 |
90.5 |
|
Distant lymph node(s) |
0 |
0 |
|
Distant metastasis except distant lymph node(s), Carcinomatosis |
31 |
5.2 |
|
Distant metastasis plus distant lymph nodes |
7 |
1.2 |
|
Unknown |
19 |
3.2 |
|
Location of Metastases |
|
|
|
Bone |
2 |
0.3 |
|
Lung |
17 |
2.8 |
|
Liver |
0 |
0 |
|
Brain |
0 |
0 |
|
Lymphatic or Vascular Invasion |
11 |
1.8 |
|
Treatment modality |
|
|
|
Surgery |
544 |
91.1 |
|
Radiation |
350 |
58.6 |
|
Chemotherapy |
325 |
54.4 |
|
Adjuvant Therapy |
N=544 |
% of Total |
|
Surgery Only |
309 |
56.8 |
|
Surgery with Systemic Therapy |
37 |
6.8 |
|
Surgery with Radiation |
134 |
24.6 |
|
Surgery with Chemoradiation |
64 |
11.8 |
Table 4: 1-, 3-, 5- and 10-Year Survival Probabilities for Primary Site, Age, Sex, Grade, Stage, Histology, Tumor Size, and Adjuvant Therapy
|
Variable |
Probability of 1-Year Survival (%) |
Probability of 3-Year Survival (%) |
Probability of 5-Year Survival (%) |
Probability of 10-Year Survival (%) |
|
Overall Survival |
98.1 |
89.3 |
85.3 |
79.5 |
|
Primary Anatomical Site |
|
|
|
|
|
Head, Face and Neck |
100 |
88.0 |
84.5 |
79.8 |
|
Lung and Thorax |
89.7 |
59.8 |
50.2 |
50.2 |
|
Trunk and Pelvis |
98.6 |
87.2 |
81.2 |
71.1 |
|
Upper Limb and Shoulder |
100 |
98.2 |
95.9 |
86.7 |
|
Lower Limb and Hip |
98.4 |
89.0 |
85.0 |
80.8 |
|
Age Group (years) |
|
|
|
|
|
0-5 |
93.8 |
93.8 |
93.8 |
62.5 |
|
6-10 |
100 |
98.6 |
98.6 |
96.0 |
|
11-15 |
99.2 |
91.4 |
87.7 |
82.3 |
|
16-18 |
95.9 |
82.8 |
76.8 |
71.6 |
|
Sex |
|
|
|
|
|
Male |
97.5 |
85.7 |
80.7 |
74.2 |
|
Female |
98.8 |
93.2 |
90.3 |
85.4 |
|
Grade |
|
|
|
|
|
Well Differentiated, Differentiated |
100 |
100 |
100 |
100 |
|
Moderately differentiated, moderately well differentiated, intermediate differentiation |
100 |
96.6 |
95 |
90.5 |
|
Poorly differentiated |
97.2 |
83.6 |
78.9 |
76.0 |
|
Undifferentiated, anaplastic |
95.3 |
77.2 |
70.6 |
61.7 |
|
NCDB Analytical Stage |
|
|
|
|
|
Stage I |
100 |
95.2 |
91.7 |
90.1 |
|
Stage II |
100 |
98.1 |
93.7 |
79.2 |
|
Stage III |
97.8 |
85.6 |
82.2 |
72.2 |
|
Stage IV |
87.0 |
31.3 |
27.4 |
27.4 |
|
Histology |
|
|
|
|
|
Synovial Sarcoma, NOS |
96.0 |
81.6 |
76.7 |
67.1 |
|
Monophasic Synovial Sarcoma |
98.9 |
90.6 |
87.7 |
82.1 |
|
Biphasic Synovial Sarcoma |
100 |
97.8 |
93.8 |
93.8 |
|
Tumor Size |
|
|
|
|
|
<5 cm |
100 |
96.5 |
95.7 |
92.6 |
|
5-10 cm |
99.4 |
90.5 |
82.6 |
73.5 |
|
>10 cm |
94.7 |
75.9 |
73.4 |
73.4 |
|
Adjuvant Therapy |
|
|
|
|
|
Surgery Only |
98.9 |
92.0 |
89.4 |
86.2 |
|
Surgery with Systemic Therapy |
100 |
93.5 |
79.7 |
79.7 |
|
Surgery with Radiation |
99.2 |
91.4 |
88.3 |
81.3 |
|
Surgery with Chemoradiation |
98.1 |
86.4 |
81.3 |
76.6 |
*NOS: Not Otherwise Specified
Table 5: Hazard Ratios for Primary Site, Age, Sex, Race, Histology, Tumor Size, Grade, Stage, and Adjuvant Therapy
|
Variable |
Hazard Ratio (95% Confidence Interval) |
P-value |
|
Primary Anatomical Site |
|
|
|
Upper Limb and Shoulder |
Reference |
|
|
Head, Face and Neck |
2.374 (0.858-6.568) |
0.096 |
|
Lung and Thorax |
10.021 (4.089-24.563) |
<0.0005 |
|
Trunk and Pelvis |
3.625 (1.520-8.646) |
0.004 |
|
Lower Limb and Hip |
2.500 (1.167-5.355) |
0.018 |
|
Age Category |
|
|
|
6-10 years |
Reference |
|
|
11-15 years |
2.290 (0.509-10.308) |
0.280 |
|
16-18 years |
4.191 (0.946-18.560) |
0.059 |
|
Sex |
|
|
|
Female |
Reference |
|
|
Male |
2.251 (1.392-3.640) |
0.001 |
|
Race |
|
|
|
Caucasian |
Reference |
|
|
African American |
1.526 (.804-2.898) |
0.196 |
|
Asian |
2.386 (1.026-5.545) |
0.043 |
|
Histology |
|
|
|
Biphasic |
Reference |
|
|
Monophasic Spindle Cell |
0.473(0.283-0.789) |
0.022 |
|
Synovial Sarcoma NOS |
5.276 (2.496-11.152) |
<0.0005 |
|
Tumor size |
|
|
|
< 5 cm |
Reference |
|
|
5-10 cm |
3.473 (1.868-6.457) |
<0.0005 |
|
> 10 cm |
6.323 (3.163-12.642) |
<0.0005 |
|
Tumor grade |
|
|
|
Moderately-differentiated |
Reference |
|
|
Poorly-differentiated |
2.082 (0.832-5.209) |
0.117 |
|
Undifferentiated, anaplastic |
3.441 (1.283-9.229) |
0.014 |
|
NCDB Analytical Stage |
|
|
|
Stage I |
Reference |
|
|
Stage II |
1.091 (0.449-2.651) |
0.847 |
|
Stage III |
2.313 (1.004-5.331) |
0.049 |
|
Stage IV |
15.062 (6.526-34.761) |
<0.0005 |
|
Adjuvant Therapy |
|
|
|
Surgery Only |
Reference |
|
|
Surgery with Systemic Therapy |
1.539 (.595-3.982) |
0.374 |
|
Surgery with Radiation |
1.345 (.759-2.381) |
0.310 |
|
Surgery with Chemoradiation |
1.775 (0.864-3.647) |
0.118 |
References
- Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M et al. (2009) Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology and End Results Program, 1983 to 2005: an analysis of 1268 patients. Cancer 115: 3537-3547. [Crossref]
- Sangkhathat S (2015) Current management of pediatric soft tissue sarcomas. World J Clin Pediatr 4: 94-105. [Crossref]
- Moberger G, Nilsonne U, Friberg S (1968) Synovial sarcoma. Acta Orthop Scand 111: 3-38.
- Roth JA, Enzinger FM, Tannenbaum M (1975) Synovial sarcoma of the neck: a follow-up study of 24 cases. Cancer 35: 1243-1253. [Crossref]
- Pack GT, Ariel IM (1950) Synovial sarcoma (malignant synovioma): a report of 60 cases. Surgery 28: 1047-1084. [Crossref]
- Zhang WD, Guan YB, Chen YF, Li CX (2012) CT imaging of primary pleuropulmonary synovial sarcoma. Clin Radiol 67: 884-888. [Crossref]
- Wright PH, Sim FH, Soule EH, Taylor WF (1982) Synovial sarcoma. J Bone Joint Surg 64: 112-122. [Crossref]
- Fetsch JF, Meis JM (1993) Synovial sarcoma of the abdominal wall. Cancer 72: 469-477. [Crossref]
- Vora A, Schneider B (2013) Synovial sarcoma of the lung in a patient who received radioactive iodine therapy for thyroid cancer. Thyroid 23: 371-375. [Crossref]
- Deshmukh R, Mankin HJ, Singer S (2004) Synovial sarcoma: the importance of size and location for survival. Clin orthop Relat Res 155-161. [Crossref]
- Bergh P, Meis-Kindblom JM, Gherlinzoni F, Berlin O, Bacchini P et al. (1999) Synovial sarcoma: Identification of low and high risk groups. Cancer 85: 2596-2607. [Crossref]
- Ferrari A, Bisogno G, Alaggio R, Cecchetto G, Collini P et al. (2008) Synovial sarcoma of children and adolescents: The prognostic role of axial sites. Eur J Cancer 44: 1202-1209. [Crossref]
- Bianchi G, Sambri A, Righi A, Dei Tos AP, Picci P et al. (2017) Histology and grading are important prognostic factors in synovial sarcoma. Eur J Surg oncol 43: 1733-1739. [Crossref]
- Guillou L, Benhattar J, Bonichon F, Gallagher G, Terrier P, et al. (2004) Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22: 4040-4050. [Crossref]
- Participant User Files (2017) American College of Surgeons.
- Goldblum J, Enzinger F, Folpe A, Weiss S (2014) Enzinger and Weiss's soft tissue tumors.
- Okcu MF, Munsell M, Treuner J, Mattke A, Pappo A, et al. (2003) Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21: 1602-1611. [Crossref]
- Ferrari A, De Salvo GL, Brennan B, van Noesel MM, De Paoli A et al. (2015) Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26: 567-572. [Crossref]
- United States Census (2018) United States Government.
- Song S, Kim S, Park J, Kim HJ, Kim IH et al. (2014) Effects of Adjuvant Radiation Therapy in Patients with Synovial Sarcoma. Int J Radiation Oncol 90: S755.
- Vining CC, Sinnamon AJ, Ecker BL, Kelz RR, Fraker DL et al. (2017) Adjuvant chemotherapy in resectable synovial sarcoma. J Surg Oncol 116: 550-558. [Crossref]