Subependymal Giant Cell Astrocytoma without Clinically or Genetically Identifiable Tuberous Sclerosis Complex: A Case Report
Subependymal Giant Cell Astrocytoma without Clinically or Genetically Identifiable Tuberous Sclerosis Complex: A Case Report
A B S T R A C T
Background: Subependymal giant cell astrocytoma (SEGA) is a World Health Organization grade I tumor most commonly located in the lateral ventricles near the foramen of Monro. They are primarily associated with tuberous sclerosis complex (TSC), an autosomal dominant inherited condition that leads to a variety of tumors throughout the body. TSC must be diagnosed via either genetic criteria or clinical criteria. Cases of SEGA in patients without both genetically or clinically identifiable TSC are scarce, with only two instances identified of those genetically screened in the literature. We describe what we believe is only the third such case, a 22-year-old patient with no other clinical or genetic indicators for tuberous sclerosis.
Case Description: The patient is a 22-year-old Caucasian female who experienced progressive symptoms of headaches, tinnitus and visual obscuration. A magnetic resonance imaging study of the brain revealed a homogeneously enhancing mass in the right lateral ventricle with obstruction of the foramen of Monro. The patient underwent a right frontal craniotomy for transcortical-transventricular resection of the mass. Histologic examination of the tumor revealed a diagnosis of SEGA. Germline genetic tests for both TSC1 and TSC2 were negative, and no additional clinical features were present. The patient was thus not felt to meet the criteria for a diagnosis of TSC based on the absence of clinical features, family history, and negative genetic testing.
Conclusion: Although a rare phenomenon, SEGA should remain on the differential diagnosis for patients with newly found periventricular tumors and absence of TSC clinical features.
Keywords
SEGA, subependymal giant cell astrocytoma, tuberous sclerosis complex, TSC, case report
Introduction
Subependymal giant cell astrocytoma (SEGA) is a World Health Organization grade I tumor most commonly located in the lateral ventricles near the foramen of Monro [1]. These lesions typically arise in childhood and adolescence, but rarely after the age of 25 [2]. They are almost exclusively associated with tuberous sclerosis complex (TSC), an autosomal dominant inherited condition that leads to a variety of tumors throughout the body, including in the brain, skin, kidneys and heart. According to a revised classification system released in 2012, TSC must be diagnosed via either genetic criteria (mutation of the TSC1 or TSC2 genes) or clinical criteria (two major features or one major feature with two or more minor features) [3]. Cases of SEGA in patients without both genetically or clinically identifiable TSC are scarce, with only two instances identified of those genetically screened in the literature [2, 4]. We describe what we believe is only the third such case, a 22-year-old patient with no other clinical or genetic indicators for tuberous sclerosis.
Case Description
The patient is a 22-year-old Caucasian female who experienced progressive symptoms of headaches, tinnitus and visual obscuration. A magnetic resonance image (MRI) study of the brain revealed a homogeneously enhancing mass in the right lateral ventricle with obstruction of the foramen of Monro and dilation of the right lateral ventricle consistent with obstructive hydrocephalus (Figure 1). No additional lesions were noted intracranially, and the patient had no history of cognitive disability or epilepsy. Physical exam was notable for the presence of papilledema. No dermatologic abnormalities were noted except for hypomelanotic macules. Family history was nonsignificant. The patient underwent a right frontal craniotomy for transcortical-transventricular resection of the mass. At the time of surgery, the tumor was observed to arise primarily from the floor of the lateral ventricle and was densely adherent to the thalamostriate vein in the region of the foramen of Monro. A near-total surgical resection of the lesion was performed, leaving a small residual area to prevent venous injury (Figure 2).
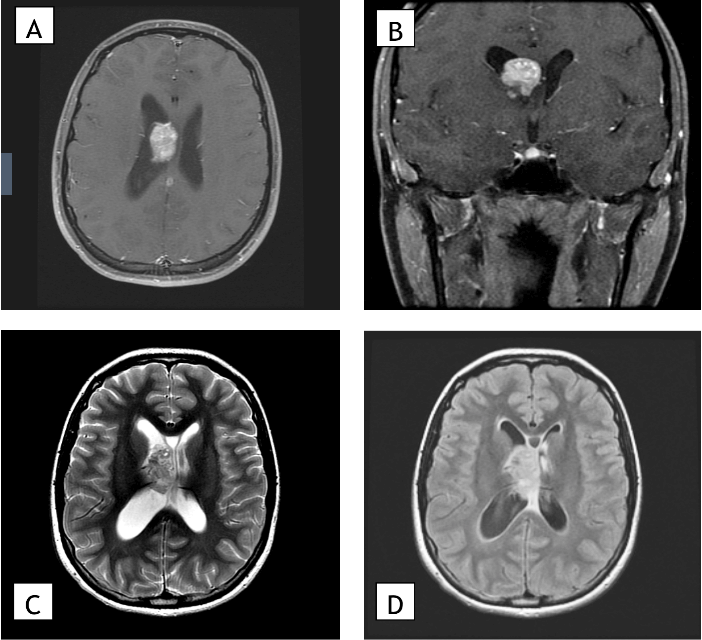
Figure 1: Preoperative MRI. A) Preoperative magnetic resonance images showing an axial section and B) coronal section of a T1 post-gadolinium contrast sequence, C) an axial section of a T2 sequence, and D) an axial section of a FLAIR sequence. Imaging shows an enhancing 3.7 cm mass in the right lateral ventricle with associated mild hydrocephalus.
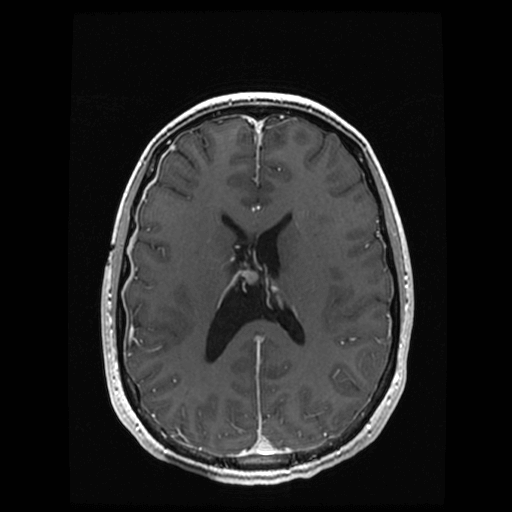
Figure 2: Postoperative follow-up MRI. Postoperative magnetic resonance images showing axial section of a T1 post-contrast sequence. Postoperative changes are seen in the region of the right foramen of Monro with resolution of the preoperative hydrocephalus.
The lesion consisted of multiple soft, red-brown tissue fragments. Microscopic examination revealed confluent sheets of tumor cells in a fibrillar background, many of which being large with pink, glassy astrocytic cytoplasm (Figure 3). Additional rare tumor cells were ganglion-like in appearance. Tumor cells were variably positive by immunohistochemistry for glial fibrillary acidic protein (GFAP), neurofilament and synaptophysin. Immunohistochemical staining for Neu-N, EMA, IDH-1 (R132H mutation), BRAF (V600E mutation) and p53 were all negative in tumor cells. Immunohistochemical stain for ATRX showed retained staining of tumor cell nuclei for ATRX, which suggested this tumor is neither a pleomorphic xanthoastrocytoma nor a diffuse glioma. Immunohistochemical staining for c-kit highlighted numerous mast cells in the vicinity of the tumor. The histologic appearance and immunohistochemical staining profile of this tumor are diagnostic of a SEGA.
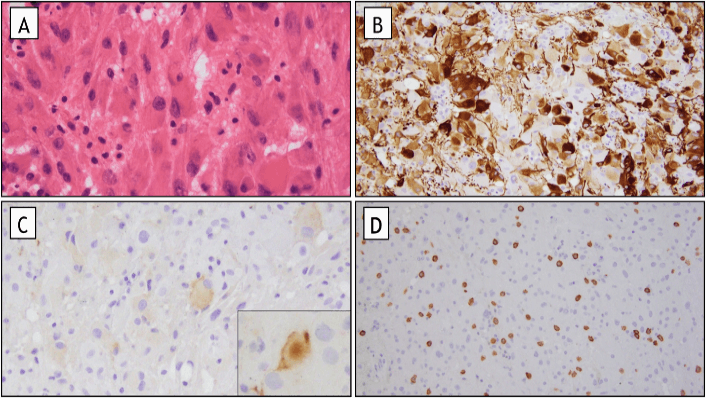
Figure 3: Pathology. A) Characteristic large ganglion like cells (H&E stain, original magnification 400X). B) Variable positive staining for GFAP with strong expression in only a subset of tumor cells (GFAP immunohistochemical stain, original magnification 200X). C) Focal expression of the neuronal marker synaptophysin in tumor cells (original magnification, including inset, 200x). D) Intratumoral mast cells, highlighted on c-kit immunohistochemical stain (c-kit immunohistochemical stain (original magnification 100X).
Due to the strong association between SEGA and TSC, the patient was referred for genetic evaluation. Germline genetic testing was performed on blood for TSC1 and TSC2, both of which were negative. She underwent bilateral renal ultrasounds to screen for angiolipomas, which were not detected. The patient was not felt to meet the criteria for a diagnosis of TSC based on the absence of additional major or minor clinical features, family history and negative genetic testing. The patient’s postoperative course was complicated by the development of intraventricular adhesions requiring ventriculoperitoneal shunt placement for hydrocephalus, and she has been followed without evidence of tumor recurrence for two years.
Discussion
We describe the case of a young female who presents with seemingly de novo SEGA formation in her right lateral ventricle without evidence of underlying tuberous sclerosis or TSC1/2 genetic abnormalities. Given the lack of syndromic features and adult age of presentation, there was a low pre-operative suspicion of her ultimate pathology of SEGA. Despite the near-universal association of SEGA with TSC, we were unable to find any additional features to satisfy the 2012 revised diagnostic criteria (Table 1) [3]. The findings of either TSC1 or TSC2 mutations are pathognomonic for TSC. A total of 2 major clinical features or 1 major and 2 minor clinical features also yields a definitive diagnosis of TSC [3]. SEGA is one example of a major diagnostic feature due to the almost exclusive association of SEGA with TSC. Of patients with TSC, approximately 5-20% have SEGA. However, almost all patients with SEGA suffer from TSC [5, 6]. Only sparse cases in the literature detail patients with SEGA and no clinical signs of TSC [7-11]. Genetic sequencing was not performed or not available in these cases, and we cannot comment on the possibility of underlying genetic mosaicism in those instances. Our patient did have genetic testing, which was negative for mutations within the TSC1/TSC2 genes and further clinical examination revealed only hypomelanotic macules of unclear age of onset, failing to fulfill minor criteria for TSC. Additional screening for renal angiolipoma, a common sequela of TSC, was also negative.
Table 1: Diagnostic criteria for tuberous sclerosis complex, as described by the 2012 International Tuberous Sclerosis Complex Consensus Conference [3]. Table adapted from Northrup & Krueger [3].
|
Genetic Diagnostic Criteria |
Clinical Diagnostic Criteria |
|
|
Major Features |
Minor Features |
|
|
TSC1 or TSC2 mutations |
Hypomelanotic macules Angiofibromas Ungual fibromas Shagreen patches Multiple retinal hamartomas Cortical dysplasias Subependymal nodules Subependymal giant cell astrocytoma Cardiac rhabdomyoma Lymphangioleiomyomatosis Angiomyolipomas |
“Confetti” skin lesions Dental enamel pits Intraoral fibromas Retinal achromic patch Multiple renal cysts Nonrenal hamartomas
|
Genetically, TSC is typically screened for using the TSC1 gene (chromosome 9a34) or the TSC2 gene (chromosome 16p13), both of which are tumor suppressor genes. These mutations are sporadic approximately 60-80% of the time [6, 12]. Genetic analysis for deletions and mutations in both TSC1 and TSC2 were performed on this patient’s peripheral blood, and no abnormalities were identified. Despite this, it must be noted that 10-25% of those with TSC have no identifiable mutation using conventional genetic testing. A possible cause for this is mosaicism. As the traditional genetic testing involves direct sequencing of the exons of the TSC1 and TSC2 genes, mutations present in leukocyte DNA can be missed [13]. This was the case in Kwiatkowska and colleagues’ case report [14]. A second possibility is an isolated somatic mutation that would be found by sequencing from the tumor itself. This was the case in a case report much like ours: a 20-year-old patient with histologically identified SEGA with no germline mutations for the aforementioned genes but had two somatic hits to the TSC2 gene when sequenced from tumor tissue [8]. An additional case report highlighted an incidental finding of SEGA in a 75-year-old female who had negative germline mutations but did have somatic mutations within the tumor itself [9]. Unfortunately, in our case, sequencing of the tumor for somatic mutations was not done.
Currently, we continue to follow our patient with repeat MRI scans. Despite a complicated immediate postoperative course, she has done very well overall, continuing to perform at a high cognitive level with complete independence. Given her clinical stability and negative testing, we have increased the duration between follow-up appointments with routine visits annually.
Conclusion
We present a case of a 22-year-old patient with SEGA in the setting of genetically or clinically negative TSC. To our knowledge, this is only the third such case in the literature. Although a rare phenomenon, SEGA should remain on the differential diagnosis for patients with newly found periventricular tumors and absence of TSC clinical features.
Funding
None.
Article Info
Article Type
Case ReportPublication history
Received: Thu 04, Jun 2020Accepted: Fri 03, Jul 2020
Published: Fri 31, Jul 2020
Copyright
© 2023 Christopher J. Farrell. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.CRSS.2020.02.03
Figures & Tables
Table 1: Diagnostic criteria for tuberous sclerosis complex, as described by the 2012 International Tuberous Sclerosis Complex Consensus Conference [3]. Table adapted from Northrup & Krueger [3].
|
Genetic Diagnostic Criteria |
Clinical Diagnostic Criteria |
|
|
Major Features |
Minor Features |
|
|
TSC1 or TSC2 mutations |
Hypomelanotic macules Angiofibromas Ungual fibromas Shagreen patches Multiple retinal hamartomas Cortical dysplasias Subependymal nodules Subependymal giant cell astrocytoma Cardiac rhabdomyoma Lymphangioleiomyomatosis Angiomyolipomas |
“Confetti” skin lesions Dental enamel pits Intraoral fibromas Retinal achromic patch Multiple renal cysts Nonrenal hamartomas
|
References
- Roth J, Roach ES, Bartels U, Jóźwiak S, Koenig MK et al. (2013) Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012. Pediatr Neurol 49: 439-444. [Crossref]
- Konakondla S, Jayarao M, Skrade J, Giannini C, Workman MJ et al. (2016) Subependymal giant cell astrocytoma in a genetically negative tuberous sclerosis complex adult: Case report. Clin Neurol Neurosurg 150: 177-180. [Crossref]
- Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group (2013) Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 49: 255-265. [Crossref]
- Beaumont TL, Godzik J, Dahiya S, Smyth MD (2015) Subependymal giant cell astrocytoma in the absence of tuberous sclerosis complex: case report. J Neurosurg Pediatr 16: 134-137. [Crossref]
- Adriaensen ME, Schaefer Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA et al. (2009) Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 16: 691-696. [Crossref]
- Tahiri Elousrouti L, Lamchahab M, Bougtoub N, Elfatemi H, Chbani L et al. (2016) Subependymal giant cell astrocytoma (SEGA): a case report and review of the literature. J Med Case Rep 10: 35. [Crossref]
- Halmagyi GM, Bignold LP, Allsop JL (1979) Recurrent subependymal giant-cell astrocytoma in the absence of tuberous sclerosis. Case report. J Neurosurg 50: 106-109. [Crossref]
- Ichikawa T, Wakisaka A, Daido S, Takao S, Tamiya T et al. (2005) A case of solitary subependymal giant cell astrocytoma: two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J Mol Diagn 7: 544-549. [Crossref]
- Takei H, Adesina AM, Powell SZ (2009) Solitary subependymal giant cell astrocytoma incidentally found at autopsy in an elderly woman without tuberous sclerosis complex. Neuropathology 29: 181-186. [Crossref]
- Prahlow JA, Teot LA, Lantz PE, Stanton CA (1995) Sudden death in epilepsy due to an isolated subependymal giant cell astrocytoma of the septum pellucidum. Am J Forensic Med Pathol 16: 30-37. [Crossref]
- Bonnin JM, Rubinstein LJ, Papasozomenos SC, Marangos PJ (1984) Subependymal giant cell astrocytoma. Significance and possible cytogenetic implications of an immunohistochemical study. Acta Neuropathol 62: 185-193. [Crossref]
- Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP et al. (2007) Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 9: 88-100. [Crossref]
- Eng C, Vijg J (1997) Genetic testing: the problems and the promise. Nat Biotechnol 15: 422-426. [Crossref]
- J Kwiatkowska, J Wigowska-Sowinska, D Napierala, R Slomski, D J Kwiatkowski (1999) Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N Engl J Med 340: 703-707. [Crossref]