
The Basis for the Development of FTX 6058
Researchers at the Fulcrum Therapeutics developed a bioavailable drug candidate called FTX 6058 – (a novel small molecular fetal haemoglobin inducer for sickle cell disease) that can restore the body’s ability to produce fetal haemoglobin, which is normally produced in only small amounts after birth. FTX-6058 was demonstrated to inhibit the novel biological target and elevate the expression of HBγ, resulting in the induction of fetal haemoglobin (HbF) tetramer in differentiated human primary CD34+ cells. FTX-6058 aims to treat patients by raising levels of fetal haemoglobin in cells.
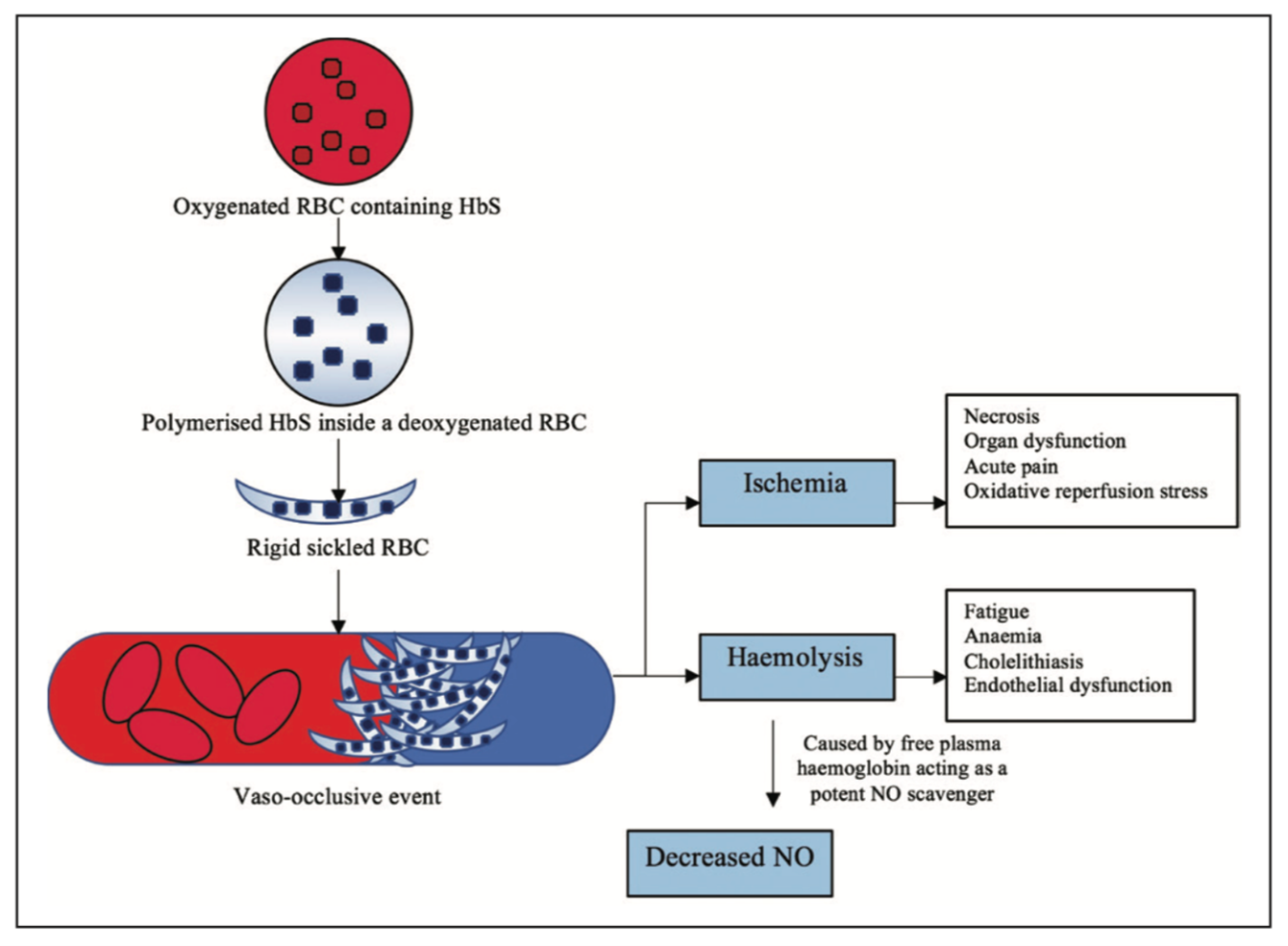
During the development, HbF is the predominant form of haemoglobin in the womb. The amount slowly decreases throughout infancy as the adult version of haemoglobin, haemoglobin A (HbA), replaces it. Researchers found out that increasing the amount of HbF — fetal haemoglobin is more effective at carrying oxygen in cells than its adult version that will help prevent the sickling of the red blood cells and relieve disease symptoms by improving blood flow and oxygen transport. Researchers targeted the fetal Hb because the fetal haemoglobin protects the red blood cells from sickling, hence infants do not show any symptoms of SCD at birth. The fetal haemoglobin gets replaced by sickle haemoglobin, when the infant is around 4 to 5 months of age, at that time the cells begin to form sickle. Upon deoxygenation, the sickle haemoglobin is insoluble and undergoes polymerization and aggregation of the polymers into tubulin fibers that cause sickling.
FTX-6058 Shows Promising Efficacy as SCD Therapy in Disease Models
FTX-6058 is a highly potent small-molecule inhibitor of Embryonic Ectoderm Development (EED) capable of inducing robust HbF protein expression in the cell and murine models. In vivo experiments in the mouse model of SCD showed that treatment with FTX-6058 increased both the RNA and protein levels of HbF, even more than hydroxyurea, an approved medication currently used to treat sickle cell disease. Treatment with FTX-6058 was found to be specifically and selectively increase the levels of HbF, without affecting other components of haemoglobin. Scientists developed FTX-6058 as an oral therapeutic designed to induce expression of fetal haemoglobin (HbF) to compensate for the mutated adult haemoglobin in SCD, resulting in reduction or elimination of symptoms.
Reason Behind Restoration of Fetal Haemoglobin
Humans produce an alternative form of haemoglobin in the womb – called fetal haemoglobin, but the production of fetal Hb reduces shortly after birth. This reveals that people with sickle cell disease immediately do not start developing symptoms until 3 or 4 months after birth. This is called the hereditary persistence of fetal haemoglobin, which can protect people who inherit the modified gene for adult haemoglobin. With fetal haemoglobin levels at around 25-30% of usual adult haemoglobin levels, these individuals may not show any disease symptoms. The oral drugs target stem cells in the bone marrow that are destined to become the red blood cells. It inhibits a protein in the cells that generally “silences” or “switches off” the gene that produces fetal haemoglobin shortly after birth.